
Fe2O3@Au core@shell nanoparticle–graphene nanocomposites as theranostic agents for bioimaging and chemo-photothermal synergistic therapy†
Hongda Chena,
Fuyao Liuab,
Zhen Leiab,
Lina Ma*a and
Zhenxin Wang*a
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, P. R. China. E-mail: malina@ciac.ac.cn; wangzx@ciac.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100039, P. R. China
First published on 30th September 2015
Abstract
Herein, we present the synthesis and application of a new type of graphene-based magnetic and plasmonic nanocomposite for magnetic-field-assisted drug delivery and chemo-photothermal synergistic therapy. The nanocomposites were prepared via conjugation of the PEGylated Fe2O3@Au core/shell nanoparticles (Fe2O3@Au NPs) with reduced graphene oxide (rGO). The hybrid nanostructures (rGO–Fe2O3@Au NPs) are superparamagnetic and show great photothermal conversion efficiency under 808 nm near-infrared (NIR) laser irradiation and high drug loading ability (1.0 mg mg−1). MTT cell viability assay demonstrates that the chemotherapeutic drug, doxorubicin loaded rGO–Fe2O3@Au NPs (DOX–rGO–Fe2O3@Au NPs) have synergistic interaction between photothermal therapy (PTT) and chemotherapy. Furthermore, in vitro studies using HeLa cells show that the chemo-photothermal therapeutic efficacy of DOX–rGO–Fe2O3@Au NPs can be dramatically improved by the assistance of magnetic-field-guided drug delivery.
Introduction
During the last two decades, novel nanomaterials and advanced nanotechnologies continue to strengthen the development of theranostic agents/methods for efficient cancer diagnosis and therapy.1 In particular, nanomaterials can be used to integrate more than one kind of therapeutic functions for achieving combination therapy, which is usually considered as a promising strategy to improve therapeutic efficiency, reverse drug resistance, and minimize side effects.2 Recently, imaging guided chemo-photothermal therapy has attracted tremendous attention since the drug delivery and cancer therapy processes can be well monitored non-invasively by bioimaging, and mild photothermal heating could trigger the release of drug molecules from the nanocarriers to improve anticancer efficacy of chemotherapeutics by a synergistic manner.3–10A variety of nanomaterials with optical absorption in the near-infrared (NIR) spectrum including two dimension (2D) nanosheets, carbon-based nanomaterials, inorganic nanoparticles and nanoscaled conducting polymers have been used as photothermal transducing agents and drug carriers since normal tissues are transparent in the NIR region (700–1000 nm).11–16 Because graphene (graphene oxide (GO) or reduced graphene oxide (rGO)) has efficient loading capacity and superior photothermal sensitivity, graphene has been extensively employed as ideal nanoplatform to fabricate theranostic agents for chemo-photothermal therapy.17–20 For instance, Guo and coauthors have developed a doxorubicin-loaded PEGylated nanographene oxide (NGO–PEG–DOX) nanocomposite for chemo-photothermal therapy.21 The chemo-photothermal treatment based on NGO–PEG–DOX is superior to chemotherapy or photothermal treatment alone, resulting in high therapeutic efficacy and low systematic toxicity. In addition, iron oxide@gold core@shell and/or core@satellites nanoparticles FexOy@Au NPs (x = 2 or 3 and y = 3 or 4) are known to have a wide range of promising applications in bioanalytical and biomedical fields because the FexOy@Au NPs can offer facile functionalization, enhanced chemical stability, excellent biocompatibility and super-paramagnetic capability.22–36 In particular, the FexOy@Au NPs can serve as magnetic resonance imaging (MRI)-capable and NIR-resonant nanomediators for developing MRI-guided photothermal therapy (PTT).22–26 Recently, Liu and coauthors have demonstrated that iron oxide nanoparticles (IONPs) and Au NPs decorated GO (GO–IONP–Au) nanocomposites could serve as a powerful theranostic agents for in vitro PTT cancer cell killing under molecular targeting or magnetic targeting or in vivo dual model-imaging guided photothermal tumor destruction.37 However, there are few of examples on development of graphene–FexOy@Au NPs nanocomposites for biomedical applications.
In this work, we proposed a covalent method to prepare nanocomposites of rGO and Fe2O3@Au core/shell NPs, which are then used as carriers for loading doxorubicin hydrochloride (DOX), obtaining DOX–rGO–Fe2O3@Au NPs. The as-prepared DOX–rGO–Fe2O3@Au nanocomposites exhibit good magnetic property, high photothermal conversion efficiency and light-triggered DOX release. In vitro studies have demonstrated that the chemo-photothermal therapy with magnetic attraction is more effective than chemo-photothermal therapy without magnetic attraction, PTT or chemotherapy alone.
Experimental sections
Materials
Chloroauric acid (HAuCl4·3H2O, 99.99%), and PEG–SH (O-[2-(3-mercaptopropionylamino) ethylmercaptoethyl]-O′-methylpolyethyleneglycol, MW = 5000) were obtained from Sigma-Aldrich Co. (St Louis, USA). Amino-modified PEG–SH (NH2–PEG–SH, MW = 2000) was obtained from Nanocs Inc. (Boston, USA). Graphite oxide was obtained from Nanjing XFNANO Materials Tech Co., Ltd, (Nanjing, China). N-Hydroxysulfosuccinimide (sulfo-NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) were obtained from Alfa Aesar (Ward Hill, USA). HeLa cell line was purchased from Shanghai Cell Bank (Shanghai, China). Dulbecco's modide Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco Co. (New York, USA). Doxorubicin hydrochloride (DOX) was purchased from Sangon Ltd. (Shanghai, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was received from Beijing Dingguo Biotechnology Ltd. (Beijing, China). Calcein AM and propidium iodide (PI) were purchased from Aladdin Reagent Co. (Shanghai, China). Other Chemicals were analytical grade and obtained from Beijing Chemical Reagents Co. (Beijing, China). Milli-Q water (18.2 MΩ cm) was used in all experiments.Characterization
Transmission electron microscope (TEM) micrographs were performed on a JEM 2000FX (Jeol Ltd, Japan), energy-dispersive X-ray spectra (EDS) were inspected on an energy dispersive spectroscopy (FEI Co., USA). Dynamic light scattering (DLS) and zeta potential distribution measurements were carried out on Malvern Zetasizer Nano ZS (Malvern Instruments Ltd., UK). UV-visible spectra were recorded using a Mini 1240 UV-visible spectrophotometer (Shimadzu Co., Japan). Magnetization measurements were characterized by Lakeshore-7410 vibrating sample magnetometry (VSM, Lakeshore, USA). The T2-weighted magnetic resonance (MR) images were acquired using a Siemens 1.5 T MRI scanner (Magnetom Avanto, Siemens Co., Erlangen, Germany). Raman spectra were carried out on a J-Y T64000 Raman spectrometer (Horiba Jobin-Yvon Co., France).Preparation of amino functionalized PEGylated Fe2O3@Au NPs
The Fe2O3@Au NPs were synthesized according to the previously reported protocol with slight modification.38,39 Briefly, 1.08 g FeCl3 and 0.4 g FeCl2 were successively dissolved in 5 mL HCl (0.5 M) solution. The solution was added dropwisely into 50 mL NaOH (1.5 M) solution with vigorous stirring. Then, the black Fe3O4 precipitate was collected and washed by H2O and 0.01 M HCl until the solution pH value changed to 6. The fresh prepared Fe3O4 NPs were dissolved in 0.01 M HNO3 and incubated at 100 °C for 1 h under vigorous stirring. Subsequently, the as-prepared γ-Fe2O3 NPs were collected and washed by 50 mL H2O (2 times). In the presence of 5 mM sodium citrate and 2 mM hydroxylamine, gold atoms were sequentially deposited onto the γ-Fe2O3 NPs (2.2 mM as Fe content) via reduction of 1% AuCl4− (525 μL). After purified with repeated centrifugation (6000 rpm for 10 min, 3 times), the Fe2O3@Au NPs were reacted with NH2–PEG–SH at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 molar ratio (Fe2O3@Au NP:NH2–PEG–SH) for 12 h, and then PEG–SH were added into the reaction mixture and incubated for another 4 h, respectively. The final molar ratio of Fe2O3@Au NP to PEG–SH was 1:5000 in the reaction mixture. After removed excess NH2–PEG–SH and PEG–SH by centrifugation (6000 rpm for 10 min, 3 times), the as-prepared amino functionalized PEGylated Fe2O3@Au NPs (termed as NH2–PEG–Fe2O3@Au NPs) were redispersed in H2O and stored at 4 °C.
:500 molar ratio (Fe2O3@Au NP:NH2–PEG–SH) for 12 h, and then PEG–SH were added into the reaction mixture and incubated for another 4 h, respectively. The final molar ratio of Fe2O3@Au NP to PEG–SH was 1:5000 in the reaction mixture. After removed excess NH2–PEG–SH and PEG–SH by centrifugation (6000 rpm for 10 min, 3 times), the as-prepared amino functionalized PEGylated Fe2O3@Au NPs (termed as NH2–PEG–Fe2O3@Au NPs) were redispersed in H2O and stored at 4 °C.
Preparation of carboxylated rGO
The rGO was prepared by literature reported methods.40 Generally, 10 mL graphite oxide (2 mg mL−1) were ultrasonicated at room temperature for 2 h. The graphene oxide (GO) dispersion was purified by centrifugation (9000 rpm for 15 min, 2 times) and the brown supernatant was retained and diluted to 20 mL by H2O. Then, 1 g ClCH2COONa and 1 g NaOH were added into the GO dispersion to convert the –OH groups into –COOH groups on the GO surface. After 2 h ultrasonication, the GO–COOH dispersion was neutralized with dilute hydrochloric acid and purified by repeated centrifugation (9000 rpm for 15 min, 3 times). Subsequently, 0.2 g NaBH4 were added into the supernatant under vigorous stirring for 10 min, followed by incubation at 90 °C for 1 h, and purified by centrifugation (9000 rpm for 15 min, 3 times). The obtained carboxylated rGO precipitants (named as rGO–COOH) were redispersed in H2O and stored at room temperature.Synthesis of rGO–Fe2O3@Au nanocomposites
The rGO–COOH (0.2 mg mL−1) were mixed with EDC and sulfo-NHS (2 mM and 5 mM) in MES (10 mM, pH 7) buffer and incubated for 20 min. NH2–PEG–Fe2O3@Au NPs were added with a final concentration of 0.1 mg mL−1, followed by stirring for another 1 h. Then, the solution was treated with NaOH (10 mM), ultrasonicated for 1 h and centrifuged (6000 rpm, 3 times) to remove physically adsorbed NH2–PEG–Fe2O3@Au NPs. Finally, the as-prepared rGO–Fe2O3@Au NPs nanocomposites (termed as rGO–Fe2O3@Au NPs) were redispersed in H2O. The surface density of Fe2O3@Au NP on rGO sheet is c.a. 270 NPs per μm2 as determined by TEM.Drug loading and releasing of rGO–Fe2O3@Au NPs
For loading of DOX onto rGO–Fe2O3@Au NPs, 1 mg rGO–Fe2O3@Au NPs were mixed with 2 mg DOX in 10 mL PBS (10 mM phosphate buffer (PB) with 137 mM NaCl, pH 7.4) for 12 h. Excess DOX were removed by repeated centrifugation (9000 rpm for 10 min, 3 times). The obtained nanocomposites were named as DOX–rGO–Fe2O3@Au NPs. In the releasing experiment, the DOX–rGO–Fe2O3@Au NP solutions were incubated in PBS at 7.4 for desired period without or with 808 nm NIR laser (2 W cm−2) irradiation for 5 min at the suitable time points (0 and 55 min), respectively. After centrifugation at 9000 rpm for 15 min, the amounts of released DOX in the supernatant solutions were measured by UV-visible spectroscopy.Cell uptake and cell viability assay
HeLa cells were cultured in DMEM supplemented with 10% (v/v) FBS and penicillium–streptomycin (100 U mL−1 penicillin, 100 U mL−1 streptomycin) under 5% CO2 at 37 °C. Cells were firstly seeded at a density of 1 × 104 cells per well into 96-well plates and cultured for 24 h. Then, the culture medium was replaced with 100 μL of fresh culture medium containing nanocomposites with desired concentrations and incubated for another 24 h. Subsequently, the cells were washed by fresh DMEM supplemented with 10% (v/v) FBS (100 μL, 3 times) and PBS (100 μL, 3 times). Cell viability was determined using the traditional MTT toxicology assay. The cell viability was expressed as the percentage of live cells over that of the control. The cells cultured under same condition except nanocomposite treatment were employed as control. To quantitatively evaluate the cellular uptake efficiency, the nanocomposite-stained cells were detached from the culture plates by 100 μL trypsin, concentrated by centrifugation (1000 rpm for 5 min), redispersed in 100 μL of PBS for absorption spectral measurements, respectively. For in vitro MR imaging, rGO–Fe2O3@Au NP-stained HeLa cells pellets with 1 × 106 cells were immobilized at the bottom of 1.5 mL Eppendorf tubes by 1% agarose, respectively. MR imaging were performed under following parameters: repetition time (TR), 4000 ms; echo time (TE), 84 ms; field of view, 102 mm × 72 mm; and slice thickness, 3.0 mm. Untreated HeLa cells were employed as control.In vitro chemo-photothermal therapy
The HeLa cells were seeded and cultured as previously described. After incubated with desired concentration of nanocomposites for 2 h, the cells were irradiated by an 808 nm NIR laser (2 W cm−2) for 5 min. Subsequently, the cells were washed by fresh DMEM supplemented with 10% (v/v) FBS (100 μL, 3 times) and PBS (100 μL, 3 times), and incubated in 100 μL fresh culture medium for another 24 h. MTT cell viability assay was conducted to determine the cell killing efficiency.The in vitro magnetic field-assisted chemo-photothermal therapy experiments were conducted by placing a magnet under the center of the cell culture dish. The HeLa cells were cultured, incubated with rGO–Fe2O3@Au NPs or DOX–rGO–Fe2O3@Au NPs at a rGO concentration of 0.05 mg mL−1 and irradiated by an 808 nm NIR laser as previously described. After washed with PBS (100 μL, 3 times), the cells were co-stained by 2 μM calcein AM and 4 μM propidium iodide (PI) in fresh culture medium for 0.5 h, respectively. After washed by PBS, the cells were imaged by a laser scanning confocal fluorescence microscope (ZEISS LSM780, Germany).
Results and discussion
Synthesis and characterization of rGO–Fe2O3@Au NPs
The rGO–Fe2O3@Au NPs were synthesized by covalent bonding of NH2–PEG–Fe2O3@Au NPs with rGO–COOH (as shown in Scheme 1). In this case, 30 ± 8 nm NH2–PEG–Fe2O3@Au NPs were used to synthesize rGO–Fe2O3@Au NPs since they have excellent stability and biocompatibility.33 In the presence of EDC and NHS, the NH2–PEG–Fe2O3@Au NPs can be easily conjugated with rGO–COOH through formation of amide bond between the amino group on NP surface and carboxyl group of rGO–COOH. As shown in Fig. 1, the Fe2O3@Au NPs were well dispersed both at the edge and on the plane of graphene sheets, indicating that the rGO–Fe2O3@Au NPs were successfully synthesized. The rGO–COOH has maximum absorption peak at 268 nm and high absorption above 300 nm region (as shown in Fig. S1, ESI†). A new absorption band at 542 nm of rGO–Fe2O3@Au NPs is ascribed to the presence of Fe2O3@Au NPs on the rGO, which also confirms the formation of rGO–Fe2O3@Au NPs (as shown in Fig. S1, ESI†). After conjugating with rGO–COOH, the surface plasmon resonance (SPR) band of Fe2O3@Au NPs is red-shifted from 535 nm to 542 nm, indicating that Fe2O3@Au NPs are anchored on the GO sheets. Both of rGO–COOH and rGO–Fe2O3@Au NPs have two Raman bands centered at about 1358 cm−1 and 1596 cm−1 (as shown in Fig. S2, ESI†), which are the disorder induced D band and G band of carbon atoms, respectively.41 Due to the surface-enhanced Raman scattering (SERS) property of Fe2O3@Au NPs, the Raman signal of the rGO–Fe2O3@Au NPs is slightly enhanced.42,43 The Zeta potentials and hydrodynamic sizes of as-prepared NH2–PEG–Fe2O3@Au NPs, rGO–COOH and rGO–Fe2O3@Au NPs were summarized in Table S1 in ESI.† The change of Zeta potential further demonstrates the formation of rGO–Fe2O3@Au NPs. In addition, the hydrodynamic size and UV-visible spectrum of rGO–Fe2O3@Au NPs do not show any significant change when the rGO–Fe2O3@Au NPs were incubated in fresh DMEM supplemented with 10% (v/v) FBS for 5 h. The experiment results indicate that the as-prepared rGO–Fe2O3@Au NPs have good colloidal stability (as shown in Fig. S3, ESI†).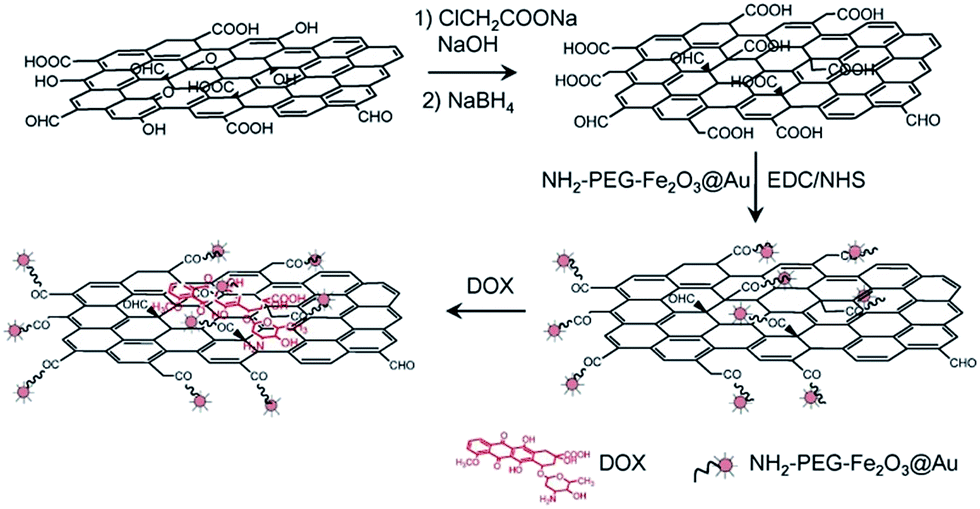 | ||
| Scheme 1 Schematic representation of preparing DOX–rGO–Fe2O3@Au NPs. The illustration is not drawn to scale. | ||
 | ||
| Fig. 1 TEM micrographs of (a) NH2–PEG–Fe2O3@Au NPs, (b) rGO–COOH and different magnification (c and d) of rGO–Fe2O3@Au NPs. | ||
Magnetic and photothermal performances of rGO–Fe2O3@Au NPs
The rGO–Fe2O3@Au NPs can uniformly disperse in H2O, forming a stable suspension, and they are capable of magnetic separation using an external bar magnet (as shown in the up inset of Fig. 2). The superparamagnetic nature of the as-prepared rGO–Fe2O3@Au NPs is confirmed by the sigmoidal anhysteretic magnetization curve with lack of coercivity and remanence in magnetization. Because of the diamagnetic behaviour of carbon, the saturation magnetization (Ms) value of rGO–Fe2O3@Au NPs (6.37 emu g−1 at 25 °C Fe2O3) is slightly lower than that of NH2–PEG–Fe2O3@Au NPs (10.02 emu g−1 at 25 °C Fe2O3) (as shown in Fig. 2). In addition, T2-weighted MR imaging was performed to further demonstrate the magnetic property of rGO–Fe2O3@Au NPs. A noticeable darkening (with decreasing signal intensity) and thereby negative contrast was observed with increasing the concentration of rGO–Fe2O3@Au NPs (as shown in bottom inset (i) of Fig. 2). The molar relaxivity (r2) (which corresponds to the slope of the line in Fig. S4, ESI†) is estimated to be 54.99 mM−1 S−1. The experimental result suggests that the rGO–Fe2O3@Au NPs can be used as a MR contrast agent. | ||
| Fig. 2 Magnetization curves of NH2–PEG–Fe2O3@Au NPs (black dot) and rGO–Fe2O3@Au NPs (red triangle). Up inset, rGO–Fe2O3@Au NPs separated by an externally placed bar magnet, and bottom inset, (i) MR images of 0, 5, 10, 25 and 50 μg mL−1 rGO–Fe2O3@Au NPs, and (ii) rGO–Fe2O3@Au NP-stained HeLa cells which were co-cultured with 0, 10, 25, and 50 μg mL−1 rGO–Fe2O3@Au NPs, respectively. | ||
As shown in Fig. 3a, the temperature is increased by 30 °C when the rGO–Fe2O3@Au NPs solution has been irradiated by 808 nm NIR laser for 510 s and do not change significantly with further irradiation. The change of temperature reaches a maximum because of the equilibrium between the heat input and output. Under same irradiation conditions, the maximum temperature change (30 °C) of rGO–Fe2O3@Au NPs solution is larger than those of rGO–COOH solution (22 °C) and NH2–PEG–Fe2O3@Au NPs solution (14 °C). The experimental result indicates that the photothermal conversion capability of rGO–Fe2O3@Au NPs is higher than those of rGO–COOH and NH2–PEG–Fe2O3@Au NPs alone. The high photothermal conversion efficiency may due to the coupling of the absorption of rGO and SPR band of Fe2O3@Au NPs. In addition, the temperature change of rGO–Fe2O3@Au NPs solution exhibits a dose-dependent and irradiation intensity-dependent manner (as shown in Fig. 3b and c). The rGO–Fe2O3@Au NPs have combined photothermal and magnetic properties and thus offer new capabilities in biomedical diagnostics and treatment.
 | ||
| Fig. 3 Solution temperature changes of (a) different nanomaterials, (b) different concentrations of rGO–Fe2O3@Au NPs and (c) different light intensities of 808 nm NIR laser over a period of 10 min irradiation. The solution temperatures were measured every 30 s using a thermometer. (a) The concentrations of NH2–PEG–Fe2O3@Au NPs, rGO–COOH and rGO–Fe2O3@Au NPs are 15, 35 and 50 μg mL−1, respectively. (a) and (b) are irradiated by 2 W cm−2 of 808 nm NIR laser. And (c) the concentration of rGO–Fe2O3@Au NPs is 50 μg mL−1 at a rGO concentration. | ||
NIR light-triggered DOX release
For constructing multifunctional drug system, DOX, a commonly used aromatic chemotherapy drug, is mixed with rGO–Fe2O3@Au NPs. As expected, the DOX–rGO–Fe2O3@Au NPs are formed after incubated with rGO–Fe2O3@Au NPs with DOX. After loaded on the rGO–Fe2O3@Au NPs, the absorption peak of DOX is slightly red-shifted (from 478 nm to 485 nm), which shows π–π stacking interaction between DOX molecule and rGO sheet (as shown in Fig. S5, ESI†).44 The saturated maximal DOX loading efficiency of rGO–Fe2O3@Au NPs is estimated to be about 1.0 mg DOX mg−1 rGO–Fe2O3@Au NPs. The drug releasing behaviors of DOX–rGO–Fe2O3@Au NPs under pH 7.4 were investigated (as shown in Fig. 4). About 17% of DOX is released from the DOX–rGO–Fe2O3@Au NPs within 2 h. The sustained release property of our nanocomposite is consistent with many previously reported π–π stacking-based drug controlled systems.6,45–48 After irradiated under an 808 nm NIR laser (2 W cm−2, 5 min for each pulse), a burst release of DOX was observed from DOX–rGO–Fe2O3@Au NPs. Within 24 h, the NIR-triggered DOX releasing amount (55%) is about twofold higher than that of DOX releasing amount (26%) without NIR irradiation (as shown in Fig. S6, ESI†). The NIR-triggered release processes could be used to adjust the amount of intracellular drug and improve therapeutic effect. | ||
| Fig. 4 DOX release from rGO–Fe2O3@Au NPs over time in PBS. In the NIR-triggered release of DOX from rGO–Fe2O3@Au NPs, which were irradiated with an 808 nm NIR laser (2 W cm−2) for 5 min at time points indicated by arrows. Error bars mean standard deviations (n = 5). | ||
The cellular uptaking and cytotoxicities of rGO–Fe2O3@Au NPs and DOX–rGO–Fe2O3@Au NPs
The HeLa cells were incubated with rGO–Fe2O3@Au NPs and DOX–rGO–Fe2O3@Au NPs for 1, 2, 4, 12, and 24 h, respectively. Because the rGO–Fe2O3@Au NPs has strong SPR band at 542 nm, the cellular uptaken amounts of rGO–Fe2O3@Au NPs can be easily estimated by measurement of the absorption of rGO–Fe2O3@Au NP stained cells. The cellular uptaken amounts of rGO–Fe2O3@Au NPs and DOX–rGO–Fe2O3@Au NPs are increased dramatically within 0 to 4 h after addition of nanocomposites, indicating that both of rGO–Fe2O3@Au NPs and DOX–rGO–Fe2O3@Au NPs can be rapidly uptaken by cells (as shown in Fig. S7, ESI†). In addition, the MRI images of rGO–Fe2O3@Au NPs stained cells show significantly decreased signal intensity with increasing the concentration of rGO–Fe2O3@Au NPs in culture medium (as shown in bottom inset (ii) of Fig. 2). The result also demonstrates that rGO–Fe2O3@Au NPs are uptaken by the cells.MTT assay of HeLa cell is employed to evaluate the cytotoxicities of as-prepared DOX–rGO–Fe2O3@Au NPs and rGO–Fe2O3@Au NPs. The cell viability of rGO–Fe2O3@Au NP-stained HeLa cells is still remained above 90% when the rGO–Fe2O3@Au NPs is as high as 50 μg mL−1 at a rGO concentration in culture medium (as shown in Fig. S8, ESI†). The result confirms that the rGO–Fe2O3@Au NPs exhibit low cytotoxicity. The cell viability is decreased by increasing the concentration of DOX–rGO–Fe2O3@Au NPs in the culture medium (as shown in Fig. S8, ESI†). The result suggests that DOX–rGO–Fe2O3@Au NPs can be used as chemotherapy agents.
Magnetic field-guided chemo-photothermal synergistic therapy
Thermal effect could change the drug uptake kinetics and increase drug diffusion through increasing cell membrane permeability. Therefore, the therapeutic efficiency of DOX–rGO–Fe2O3@Au NPs could be amplified by the synergistic effect between PTT and chemotherapy. For determining whether a synergist effect exists in the combination treatment in the DOX–rGO–Fe2O3@Au NPs system, the fraction of surviving cells from the chemo-photothermal therapy, fchem–PTT, is compared with the fraction of surviving cells by additive interaction of PTT and chemotherapy, fadd (the fadd = fchem × fPTT, fPTT is the fraction of surviving cells resulting from PTT treatment, and fchem is the fraction of surviving cells resulting from chemotherapy). fchem–PTT < fadd means a synergistic effect between the two treatments, fchem–PTT = fadd means an additive effect, and fchem–PTT > fadd means an antagonistic effect.49 As shown in Fig. 5 and Table S2 in ESI,† the fchem–PTT is lower than the calculated fadd in HeLa cells in all four doses, indicating a synergist effect between chemotherapy and PTT in this cell line. In addition, the PTT efficiency of rGO–Fe2O3@Au NPs is higher than that of rGO–COOH. The result is consistent with previous experiment of photo-thermal conversion efficiency.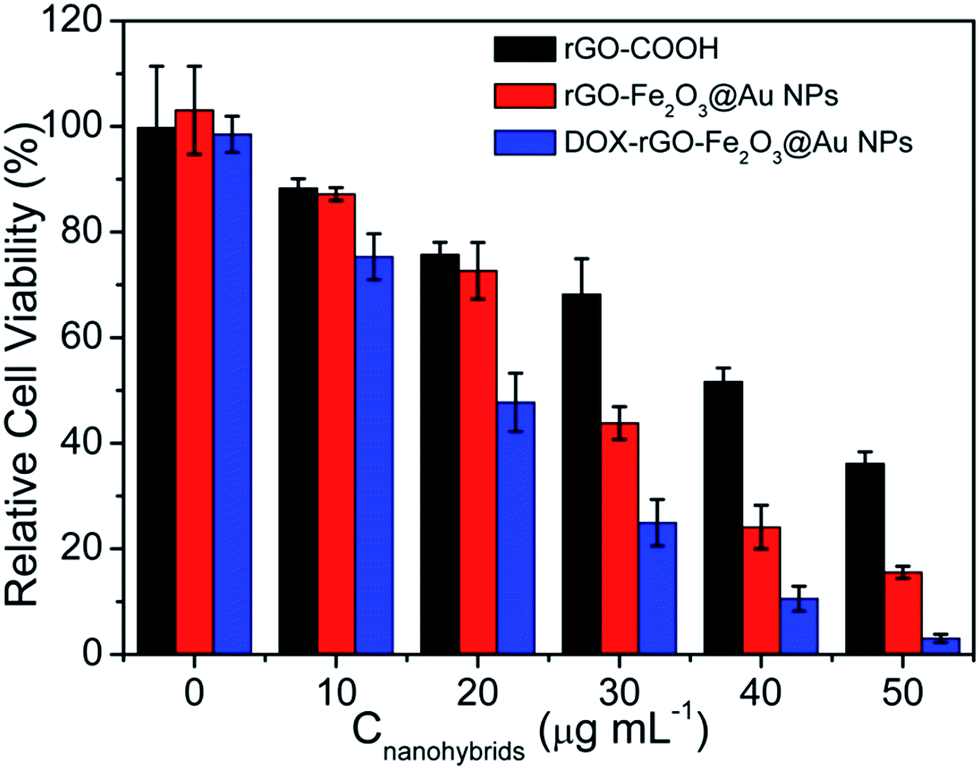 | ||
| Fig. 5 Cell viabilities of HeLa cells incubated with various concentrations of rGO–COOH, rGO–Fe2O3@Au NPs and DOX–rGO–Fe2O3@Au NPs with NIR laser irradiation (808 nm, 2 W cm−2, 5 min). The concentrations of nanohybrids are defined by rGO content. The error bars mean standard deviations (n = 5). | ||
To examine the effect of magnetic-field-guided drug delivery on the therapeutic efficacy, the magnet was placed under the cell culture plate before the addition of DOX–rGO–Fe2O3@Au NPs and remained under the plates during the whole course of experiment (as shown in the Fig. S9, ESI†). There is an apparent local accumulation of added DOX–rGO–Fe2O3@Au NPs or rGO–Fe2O3@Au NPs guided by the magnetic field. After incubation for 24 h with or without NIR laser irradiation, the HeLa cells were co-stained by calcine AM and PI and imaged by fluorescence microscopy, respectively (as shown in Fig. 6). Generally, the magnetically guided chemotherapy/chemo-thermal therapy of DOX–rGO–Fe2O3@Au NPs and PTT of rGO–Fe2O3@Au NPs are able to selectively kill cells that are localized close to the magnet, without affecting the viability of cells outside the magnetic field (as shown in Fig. 6). The phenomenon is consistent with previously literature reported GO–IONP–PEG–DOX system.11 In this magnetically guided experiment, the cell viability is followed the order, DOX–rGO–Fe2O3@Au NPs treated cells > rGO–Fe2O3@Au NPs treated cells with NIR laser irradiation > DOX–rGO–Fe2O3@Au NPs treated cells with NIR laser irradiation, which further confirms the chemophotothermal synergistic therapy of DOX–rGO–Fe2O3@Au NPs. The experimental result suggests that DOX–rGO–Fe2O3@Au NPs will be able to accumulate into tumor tissue via applying a magnetic field on the surface of tumors.
 | ||
| Fig. 6 Fluorescence images of Calcein AM/PI co-stained HeLa cells. Living cells are green and dead cells are red. (a to c) The cells were incubated with rGO–Fe2O3@Au NPs at a rGO concentration of 50 μg mL−1 for 24 h, and irradiated by 808 nm NIR laser (2 W cm−2) for 5 min. (d–i) The cells were incubated with DOX–rGO–Fe2O3@Au NPs at a rGO concentration of 50 μg mL−1 for 24 h (d–f), and irradiated by 808 nm NIR laser (2 W cm−2) for 5 min (g–i), respectively. The images were acquired at position 1 (a, d and g), position 2 (b, e and h) and position 3 (c, f and g), respectively. The details of positions are shown in the Fig. S9 in ESI.† The scale bars are 100 μm. | ||
Conclusions
In summary, the rGO–Fe2O3@Au NP nanocomposites have been successfully prepared by covalent conjugation of carboxylated rGO with amino functionalized PEGylated Fe2O3@Au NPs. The rGO–Fe2O3@Au NPs are superparamagnetic and can be employed as both high photothermal conversion agent and aromatic anticancer drug carrier. The in vitro experiment demonstrates that the DOX loaded rGO–Fe2O3@Au NPs have excellent capability for magnetic-field-guided drug delivery and chemo-photothermal synergistic therapy. The all-in-one nanoplatform paves a straightforward route to develop multifunctional theranostic agents for magnetic-guided chemo-photothermal synergistic therapy of cancer.Acknowledgements
This research was supported by National Basic Research Program of China (Grant No. 2011CB935800).Notes and references
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- E. K. Lim, T. Kim, S. Paik, S. Haam, Y. M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS PubMed.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Nano Res., 2014, 114, 10869–10939 CAS.
- A. Topete, D. Melgar, M. Alatorre-Meda, P. Iglesias, B. Argibay, S. Vidawati, S. Barbosa, J. A. Costoya, P. Taboada and V. Mosquera, J. Mater. Chem. B, 2014, 2, 6967–6977 RSC.
- M. B. Zheng, C. X. Yue, Y. F. Ma, P. Gong, P. F. Zhao, C. F. Zheng, Z. H. Sheng, P. F. Zhang, Z. H. Wang and L. T. Cai, ACS Nano, 2013, 7, 2056–2067 CrossRef CAS PubMed.
- F. Liu, X. He, Z. Lei, L. Liu, J. Zhang, H. You, H. Zhang and Z. Wang, Adv. Healthcare Mater., 2015, 4, 559–568 CrossRef CAS PubMed.
- Y. J. Lu, C. W. Lin, H. W. Yang, K. J. Lin, S. P. Wey, C. L. Sun, K. C. Wei, T. C. Yen, C. I. Lin, C. C. M. Ma and J. P. Chen, Carbon, 2014, 74, 83–95 CrossRef CAS PubMed.
- Y. Wang, K. Y. Wang, J. F. Zhao, X. G. Liu, J. Bu, X. Y. Yan and R. Q. Huang, J. Am. Chem. Soc., 2013, 135, 4799–4804 CrossRef CAS PubMed.
- H. Park, J. Yang, J. Lee, S. Haam, I. H. Choi and K. H. Yoo, ACS Nano, 2009, 3, 2919–2926 CrossRef CAS PubMed.
- J. W. Li, Z. L. Lyv, Y. L. Li, H. Liu, J. K. Wang, W. J. Zhan, H. Chen, H. B. Chen and X. M. Li, Biomaterials, 2015, 51, 12–21 CrossRef CAS PubMed.
- X. X. Ma, H. Q. Tao, K. Yang, L. Z. Feng, L. Cheng, X. Z. Shi, Y. G. Li, L. Guo and Z. Liu, Nano Res., 2012, 5, 199–212 CrossRef CAS.
- D.-K. Lim, A. Barhoumi, R. G. Wylie, G. Reznor, R. S. Langer and D. S. Kohane, Nano Lett., 2013, 13, 4075–4079 CrossRef CAS PubMed.
- H. Moon, D. Kumar, H. Kim, C. Sim, J.-H. Chang, J.-M. Kim, H. Kim and D.-K. Lim, ACS Nano, 2015, 9, 2711–2719 CrossRef CAS PubMed.
- L. Yang, Y.-T. Tseng, G. Suo, L. Chen, J. Yu, W.-J. Chiu, C.-C. Huang and C.-H. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 5097–5106 CAS.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. S. Casalongue, V. Daniel and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- B. Tian, C. Wang, S. Zhang, L. Feng and Z. Liu, ACS Nano, 2011, 5, 7000–7009 CrossRef CAS PubMed.
- X. H. Wang, Q. S. Han, N. Yu, J. Y. Li, L. Yang, R. Yang and C. Wang, J. Mater. Chem. B, 2015, 3, 4036–4042 RSC.
- H. Kim, D. Lee, J. Kim, T. I. Kim and W. J. Kim, ACS Nano, 2013, 7, 6735–6746 CrossRef CAS PubMed.
- J. Shi, L. Wang, J. Zhang, R. Ma, J. Gao, Y. Liu, C. Zhang and Z. Zhang, Biomaterials, 2014, 35, 5847–5861 CrossRef CAS PubMed.
- J. Bai, Y. Liu and X. Jiang, Biomaterials, 2014, 35, 5805–5813 CrossRef CAS PubMed.
- W. Zhang, Z. Y. Guo, D. Q. Huang, Z. M. Liu, X. Guo and H. Q. Zhong, Biomaterials, 2011, 32, 8555–8561 CrossRef CAS PubMed.
- S. Bhana, G. Lin, L. Wang, H. Starring, S. R. Mishra, G. Liu and X. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 11637–11647 CAS.
- M. Zhou, B. Singhana, Y. Liu, Q. Huang, T. Mitcham, M. J. Wallace, R. J. Stafford, R. R. Bouchard and M. R. Melancon, J. Biomed. Nanotechnol., 2015, 11, 1442–1450 CrossRef CAS PubMed.
- H. Chen, X. Ren, H. J. Paholak, J. Burnett, F. Ni, X. Fang and D. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 12814–12823 CAS.
- X. J. Ji, R. P. Shao, A. M. Elliott, R. J. Stafford, E. Esparza-Coss, J. A. Bankson, G. Liang, Z. P. Luo, K. Park, J. T. Markert and C. Li, J. Phys. Chem. C, 2007, 111, 6245–6251 CAS.
- J. Li, Y. Hu, J. Yang, P. Wei, W. Sun, M. Shen, G. Zhang and X. Shi, Biomaterials, 2015, 38, 10–21 CrossRef CAS PubMed.
- W. Gao, L. Ji, L. Li, G. Cui, K. Xu, P. Li and B. Tang, Biomaterials, 2012, 33, 3710–3718 CrossRef CAS PubMed.
- T. Zhou, B. Y. Wu and D. Xing, J. Mater. Chem., 2012, 22, 470–477 RSC.
- L. Lou, K. Yu, Z. Zhang, R. Huang, J. Zhu, Y. Wang and Z. Zhu, Nano Res., 2012, 5, 272–282 CrossRef CAS.
- H. D. Cai, K. G. Li, M. W. Shen, S. H. Wen, Y. Luo, C. Peng, G. X. Zhang and X. Y. Shi, J. Mater. Chem., 2012, 22, 15110–15120 RSC.
- M. Ye, Z. Wei, F. Hu, J. Wang, G. Ge, Z. Hu, M. Shao, S. T. Lee and J. Liu, Nanoscale, 2015, 7, 13427–13437 RSC.
- K. C. Leung, S. Xuan, X. Zhu, D. Wang, C. P. Chak, S. F. Lee, W. K. Ho and B. C. Chung, Chem. Soc. Rev., 2012, 41, 1911–1928 RSC.
- X. X. He, F. Y. Liu, L. Liu, T. C. Duan, H. M. Zhang and Z. X. Wanq, Mol. Pharmaceutics, 2014, 11, 738–745 CrossRef CAS PubMed.
- Z. Fan, M. Shelton, A. K. Singh, D. Senapati, S. A. Khan and P. C. Ray, ACS Nano, 2012, 6, 1065–1073 CrossRef CAS PubMed.
- L. Li, M. Nurunnabi, M. Nafiujjaman, Y. Y. Jeong, Y. K. Lee and K. M. Huh, J. Mater. Chem. B, 2014, 2, 2929–2937 RSC.
- L. Deng, S. J. Guo, Z. J. Liu, M. Zhou, D. Li, L. Liu, G. P. Li, E. K. Wang and S. J. Dong, Chem. Commun., 2010, 46, 7172–7174 RSC.
- X. Shi, H. Gong, Y. Li, C. Wang, L. Cheng and Z. Liu, Biomaterials, 2013, 34, 4786–4793 CrossRef CAS PubMed.
- L. Sun, J. Wang and Z. Wang, Nanoscale, 2010, 2, 269–276 RSC.
- J. L. Lyon, D. A. Fleming, M. B. Stone, P. Schiffer and M. E. Williams, Nano Lett., 2004, 4, 719–723 CrossRef CAS.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- Y. Y. Wang, Z. H. Ni, T. Yu, Z. X. Shen, H. M. Wang, Y. H. Wu, W. Chen and A. T. S. Wee, J. Phys. Chem. C, 2008, 112, 10637–10640 CAS.
- M. Potara, M. Baia, C. Farcau and S. Astilean, Nanotechnology, 2012, 23, 055501 CrossRef PubMed.
- Z. J. Zhu, L. Ma, M. Su, D. J. Liu and Z. X. Wang, J. Mater. Chem. B, 2013, 1, 1432–1438 RSC.
- D. Depan, J. Shah and R. D. K. Misra, Mater. Sci. Eng., C, 2011, 31, 1305–1312 CrossRef CAS PubMed.
- S. P. Sherlock, S. M. Tabakman, L. Xie and H. Dai, ACS Nano, 2011, 5, 1505–1512 CrossRef CAS PubMed.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed.
- Y. Wang, R. Huang, G. Liang, Z. Zhang, P. Zhang, S. Yu and J. Kong, Small, 2014, 10, 109–116 CrossRef CAS PubMed.
- H. Bao, Y. Pan, Y. Ping, N. G. Sahoo, T. Wu, L. Li, J. Li and L. H. Gan, Small, 2011, 7, 1569–1578 CrossRef CAS PubMed.
- T. S. Hauck, T. L. Jennings, T. Yatsenko, J. C. Kumaradas and W. C. W. Chan, Adv. Mater., 2008, 20, 3832–3838 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra17143a |
| This journal is © The Royal Society of Chemistry 2015 |