
Synthesis of well-defined α-fluorinated alkyl ester, ω-carboxyltelechelic polystyrenes and fabrication of their hydrophobic highly ordered porous films and microspheres†
Abstract
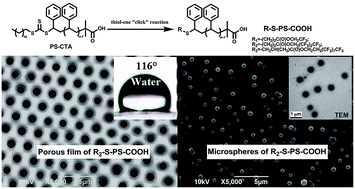
Well-defined α-fluorinated alkyl ester, ω-carboxyl telechelic polystyrenes (PS) were synthesized via combining aminolysis of RAFT-polystyrene and thiol–ene “click” reaction simultaneously. The perfluoroalkyl end groups were –CF3, –(CF2)2–CF3 and –(CF2)7–CF3, respectively. Highly ordered porous films of such telechelic PS with an average pore size about 1.00 μm were fabricated via a static breath-figure process. With the content of fluorine on the surface of porous films increasing from 1.04 wt%, to 1.64 wt% and 2.97 wt%, respectively, the water contact angle (WCA) of such porous films increased from 112° to 116° and 121° ± 0.4°, respectively, indicating their hydrophobicity. Interestingly, microspheres with average diameters of ca. 0.30–0.65 μm can be fabricated via a static BF process using methanol instead of water as the vapor atmosphere in the glass vessel.
 Please wait while we load your content...
Please wait while we load your content...

