
Nanoscale water soluble self-assembled zero-valent iron: role of stabilizers in their morphology†
Abstract
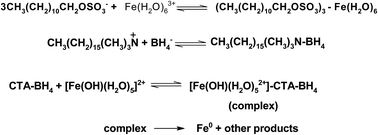
Self-assembled water soluble sheet-like zero-valent iron nano-composites have been prepared using a simple one-pot chemical reduction method using an aqueous solution of FeCl3 and NaBH4 both with and without cetyltrimethylammonium bromide (CTAB) and sodium dodecyl sulphate (SDS). It was demonstrated that dark brownish precipitates were formed immediately after the addition of NaBH4 into the ferric chloride solution. In the presence of CTAB and SDS, stable perfectly transparent brown colored iron sols were formed. UV-visible spectra show that both of the stabilizers (CTAB and SDS) formed a stable complex with Fe3+ ions. The synthesized Fe-nanoparticles were characterized using dynamic light scattering (DLS), energy dispersion X-ray spectroscopy (EDX), transmission electron microscopy (TEM), Fourier transform infrared (FT-IR) spectroscopy, ultraviolet-visible spectroscopy, and X-ray diffraction (XRD). The TEM results show that the average size of a zero-valent iron nano-sheet is about 144 to 625 nm in diameter. The mean particle size was estimated to be 203 nm with a standard deviation of 67 nm, which translated to a surface area of Fe-nanoparticles of ca. 2.0 m2 g−1. The role of surfactants and the mechanisms of nucleation and self-aggregation processes have been discussed for the first time.
 Please wait while we load your content...
Please wait while we load your content...

