DOI:
10.1039/C5RA17031A
(Paper)
RSC Adv., 2015,
5, 86102-86112
Calcination temperature-dependent surface structure and physicochemical properties of magnesium oxide†
Received
23rd August 2015
, Accepted 6th October 2015
First published on 6th October 2015
Abstract
Magnesium oxide (MgO), as an exceptionally important inorganic material, has been widely studied in view of its unique surface properties, but the correlation between its surface structure and physicochemical performance is still scarce. Here we report the evolution of the surface structure and physicochemical properties of trapezoid-like MgO microparticles with calcination temperature by transmission electron microscopy (TEM), scanning electron microscopy (SEM), energy dispersive spectroscopy (EDS), thermal gravimetric analysis (TGA) and X-ray diffraction (XRD) techniques. The results demonstrated that along with the surface change of MgO from a smooth appearance to the structure composed of nanoparticles, its corresponding crystal structure evolved from mesocrystal to polycrystal, then to pseudomorph, and finally to cubic single crystal with the increase of calcination temperature ranging from 400 °C to 1000 °C. It also illustrated that the electrochemical capability of MgO was highly dependent on its crystal structure, whereas its catalytic activity had a good correlation with its textural properties (e.g., surface area and porosity) although the reaction selectivity was related to the calcination temperature. This work highlights the vital role of calcination temperature in determining the surface structure and physicochemical properties of the inorganic material MgO, which in turn will tailor its overall performance in the final applications.
1. Introduction
Magnesium oxide (MgO) has attracted much attention owing to its outstanding properties, such as unique basicity with an isoelectric point of about 12, wide-band-gap insulativity, and special electronic and optical properties. These features have made MgO a promising candidate for applications in the fields including catalysis,1–3 adsorption and separation,4–6 chemical sensing,7,8 and electrical9 and optical10 devices. In the recent years, numerous experimental studies have demonstrated that the physicochemical properties of MgO were closely related to its surface structure,11–15 which was generally tuned by the morphologies or shapes during the synthesis process. To date, significant advances have been achieved in the synthesis of MgO using different routes including chemical vapor deposition,16 hydrothermal method,17 sol–gel technique,18 micelle-templating method,19 precursor decomposition,20 precipitation method,6,15,21 and many MgO materials with variable structures have been prepared.
Apart from exploring the strategies for synthesis, considerable efforts have been devoted to tailoring the surface properties of MgO by varying its calcination temperature. It is general knowledge that MgO is commonly obtained by heating the carbonate, basic carbonate, oxalate, hydroxide and other compounds, and hence calcination temperature would have a great impact on the surface properties of the resulting products. Based on such a case, in the earlier study, Eubank systematically investigated the properties of MgO from calcination of magnesium carbonate, basic carbonate and hydroxide at temperatures ranging from 300 °C to 2000 °C, and observed that the obtained products at lower temperatures (e.g., 300–500 °C) demonstrated a higher surface area and stronger adsorptive power in part attributable to their lattice dilation and imperfection.22 Gai et al. quantified the correlation between the catalytic reactivity and physicochemical properties of MgO from burning magnesium hydroxide-methoxide in the temperature range of 400–700 °C, in which the surface polarisability demonstrated a converse trend relative to the catalytic reactivity with increase in calcination temperature.23 Mastuli and co-workers reported that the band gap energy of MgO increased with calcination temperature.24 Pathak and Moorthy studied the influence of calcination temperature on the morphologies of MgO powders from different organic salts, and noticed that the products derived from acetate, tannate, oxinate and formate were prone to be highly sinterable with temperature, whereas the powders from oxalate, succinate and phthalate were poorly sinterable.25 To quantitatively describe the sintering level of MgO from high temperatures, a convenient graphical method for expressing the relationship between crystallite dimension and strain function was described by plotting β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ against sinθ from X-ray diffraction data.26–28 Although the above-mentioned studies are definitely favorable for gaining a deep insight into the effect of calcination temperature on the physicochemical properties of MgO, it still remains scarce how the surface structure and physicochemical properties of MgO with uniform size distribution and well-defined shape vary with temperature. As well-known, the importance of well-defined fine particles has been recognized in numerous applications, such as in ceramics, catalysis, separation, pigments, recording materials, medical diagnostics, and many others.6,29–35 For this reason, it is interesting to systematically examine the influence of calcination temperature on the surface structure and physicochemical properties of MgO.
cosθ against sinθ from X-ray diffraction data.26–28 Although the above-mentioned studies are definitely favorable for gaining a deep insight into the effect of calcination temperature on the physicochemical properties of MgO, it still remains scarce how the surface structure and physicochemical properties of MgO with uniform size distribution and well-defined shape vary with temperature. As well-known, the importance of well-defined fine particles has been recognized in numerous applications, such as in ceramics, catalysis, separation, pigments, recording materials, medical diagnostics, and many others.6,29–35 For this reason, it is interesting to systematically examine the influence of calcination temperature on the surface structure and physicochemical properties of MgO.
At an elevated temperature, the precursors for MgO (e.g., oxalate) undergo different stages from decomposition to recrystallization, and then to sintering, which are closely related to their surface properties. Herein, we provide new insights into the surface structure evolution of monodisperse MgC2O4·2H2O particles with temperature by production of MgO. Dependent on the surface structure, its physicochemical properties varied, which strongly influenced the electrochemical and catalytic performances of the resulting particles. From the present study, we can rationalize that the calcination temperature plays a significant role in affecting the surface structure and overall performance of the well-defined MgO particles.
2. Experimental
2.1. Materials
Magnesium nitrate (Mg(NO3)2·6H2O) and benzaldehyde were purchased from Tianjin Fu Chen Chemical Reagents Factory (Tianjin, China). Sodium oxalate (Na2C2O4) was from Tianjin Yongsheng Fine Chemical Co. (Tianjin, China). Absolute ethanol was from Tiajin Tianli Chemical Reagents Co. (Tianjin, China). Potassium ferricyanide (K3Fe(CN)6) was obtained from Tiajin Shengao Chemical Reagents Co. (Tianjin, China). Graphite powder was from Shanghai Jingchun Scientific Co., Ltd. (Shanghai, China). Potassium chloride and liquid paraffin was, respectively, ordered from Guangdong Guanghua Sci-Tech Co., Ltd. (Shantou, China) and Tianjin Fuyu Fine Chemical Co., Ltd (Tianjin, China).
2.2. Synthesis of trapezoid-like MgO
The procedure for preparation of trapezoid-like MgO microparticles is similar to our recent report.15 In a typical procedure, 15.38 g of Mg(NO3)2·6H2O was dissolved into 100 mL of double-deionized water, and then the Mg(NO3)2 solution was transferred into a 500 mL three-necked flask and heated to 50 °C. Subsequently, 8.04 g Na2C2O4 was added into 200 mL of solution, which was also heated to 50 °C. Under a vigorous stirring (ca. 800 rpm), the Na2C2O4 solution was poured into the Mg(NO3)2 solution in 4–5 s. The mixture was further stirred for 1 min and then maintained at the temperature of 50 °C under static conditions for 1 h. After that, a white precipitate was collected, filtered off, washed with double-deionized water and ethanol several times, and dried at 70 °C in a baking oven for 3 h. The MgO samples were prepared by calcination of the prepared trapezoid-like MgC2O4·2H2O in air from room temperature to different temperatures (400–1000 °C) in a muffle furnace followed by maintaining at the temperatures for 3 h.
2.3. Characterization
The morphology, size and energy dispersive spectroscopy (EDS) of the obtained particles were examined by a JEOL JSM-6390A scanning electron microscopy (SEM) and a FEI Tecnai G2 F20 or a JEOL JEM2100F transmission electron microscopy (TEM). The thermal decomposition property of the prepared MgC2O4·2H2O was detected by a thermal gravimetric analyzer (TGA, Mettler Toledo, TGA/DSC1 STAR System, Switzerland), which was carried out in dynamic nitrogen gas with a heating rate of 10 °C min−1. The crystal structures of as-synthesized products were characterized with X-ray diffraction (XRD) on a XRD-6000 diffractometer using Cu Kα radiation. The operation voltage was 40 kV, and the current was 30 mA. Nitrogen adsorption–desorption isotherms were performed using a Micrometrics ASAP 2020HD88 instrument at −196 °C.
2.4. Electrochemical evaluation
The cyclic voltammetric experiments were performed on a CHI 650C computer-controlled potentiostat (Chenhua Instruments Co., Shanghai, China) with a standard three-electrode system. A platinum wire was used as counter electrode, and a saturated calomel electrode (SCE, Chenhua Instruments Co., Shanghai, China) was employed as reference electrode. The working electrode was as-prepared MgO modified carbon paste (CP) electrode. The construction of MgO/CP electrode was as below: first 0.5 g graphite powder was mixed with 0.1 g liquid paraffin by a ratio of 5:1, which was filled into an electrode tube for fabrication of blank CP electrode. Then as-synthesized MgO was homogeneously mixed with the CP material same as above, and the content of MgO in the mixture was ranging from 2.5% to 7.5%, which was subsequently filled into one end of the blank CP electrode. Prior to use, the CP electrode was polished with a piece of weighting paper in order to get a smooth and uniform surface structure of MgO/CP. The electrochemical properties of the MgO/CP electrode was evaluated with the solution of potassium ferricyanide solution composed of 1 mM potassium ferricyanide and 0.1 M KCl. The voltammograms were recorded between −0.3 V and 1.0 V at a scan rate of 100 mV s−1, and the measurements were carried out in a 25 mL voltammetric cell at room temperature (ca. 25 °C).
2.5. Catalytic evaluation
The catalysts were evaluated by using the MPV reaction between ethanol and benzaldehyde. In a typical reaction, ethanol (10.5 mL, 180 mmol), benzaldehyde (0.90 mL, 9 mmol), and as-synthesized MgO (1.5 g) were transferred into a 50 mL round-bottom flask fitted with a reflux condenser. The reaction mixture was heated to the reflux temperature (78 °C) under magnetic stirring. The collected products after a reaction period of 12 h were analyzed with an Agilent GC-7890B gas chromatography equipped with a HP-INNOWax capillary column (30 m × 0.32 mm I.D., 0.25 μm film thickness).
3 Results and discussion
3.1 In situ TEM observation of the surface structure evolution of MgC2O4·2H2O
Fig. 1 shows the TEM images of an individual trapezoid-like MgC2O4·2H2O particle with extension of the arrested time. In the first 0.5 min (Fig. 1a and b), the contour of the captured particle demonstrated a polyhedron-like structure, consistent with the SEM observation in our recent study.36 In this period, the whole particle is black, which is difficult to clearly observe its surface structure. With elongation of the arrested time to around 1 min, the surface structure almost maintained except that the particle illustrated a parallelogrammic contour, and the lengths of the opposite or facing sides are 8.1 μm and 6.7 μm (Fig. 1c), respectively. This phenomenon could be ascribed to the rotation of the selected particle from one side to another side36 upon the action of high-energy convergent electron beam from TEM. Further increasing the illumination time of the accelerated electrons to around 9 min led to a great change in the surface structure of the captured particle (Fig. 1d). Apparently, the sharp edges and corners almost disappeared, and the resulting particle presented a spherical-like structure with four small corners evolved from the parallelogrammic product. More importantly, the surface structure of the obtained particle became transparent, and some regions seemed hollow and could be looked through. To the best of our knowledge, the significant change in the surface structure of the individual particle lies in the decomposition of MgC2O4·2H2O upon the illustration of high-energy convergent electron beam in the process of taking the TEM images. It is general knowledge where the beam collides with solid-state matter, electrons are converted into heat or kinetic energy.37 Under the operation condition of TEM, the electron beam provides a source of heat which leads to a rapid increase in temperature at the location of impact, quickly heating the target material. Due to the thermal instability of MgC2O4·2H2O, it will decompose into MgO, CO2 and H2O upon heating38 as described below. In the process, on the one hand, the resulting product will shrink as evidenced by the disappearance of the edges and corners of the particle, and, on the other hand, the basic composition unit of the particle will aggregate each other, leaving some hollow regions in the product. The heating effect of the electron beam was further demonstrated by extension of the illustration time. As shown in Fig. 1e, the surface structure of the product became more transparent when the illustration time was 10 min, and more interesting is that its surface structure is composed of threadlike objects. However, further prolonging the illustration time would not make the surface take a greater change (e.g., 24 min in Fig. 1f). To get a better understanding on the surface structure, the particle was examined by high resolution TEM (HRTEM). As shown in Fig. 1g, it clearly illustrates that the threadlike objects have a width of around 19 nm and a length of more than several-hundred nanometers, and also their composition is different from those particles between them. As indicated in Fig. 1h, the threadlike structures are mainly made up of some nanocrystals, whereas the components between them are primarily some amorphous particles with some nanocrystals inside (Fig. 1i). From these results, it can be speculated that upon the extensive illustration of the electron beam, the produced heat not only promotes the decomposition of MgC2O4·2H2O, but also facilitates the resulting particle recrystallize. Along the process, the produced nanocrystals would self-assemble into some threadlike structures. This assumption can be in part evidenced by the evolution of the selected area electron diffraction (SAED) patterns with time. It is obvious that the SAED patterns gradually became clear and bright, which reveals that with the decomposition of MgC2O4·2H2O, the generated amorphous particles would grow again with production of nanocrystals.
 |
| | Fig. 1 (a)–(f) In situ observation of the surface structure evolution of an individual trapezoid-like MgC2O4·2H2O particle with arrested time at (a) 0 min, (b) 0.5 min, (c) 1.0 min, (d) 9.0 min, (e) 10 min, (f) 24 min; (g) HRTEM image which shows the fine structure in (f); (h) the detailed view of the surface structure as indicated in the white circle in (g); (i) the magnification image of surface structure as indicated in the white circle in (h); (j)–(l) evolution of the selected area electron diffraction patterns with arrested times at (j) 12 min, (k) 18 min, and (l) 20 min. | |
3.2 Thermal analysis of as-synthesized MgC2O4·2H2O
In order to get an insight into the thermal decomposition behavior of as-synthesized product by the reaction between Na2C2O4 and Mg(NO3)2 at 50 °C, it was investigated by raising the temperature from 30 °C to 600 °C at a rate of 10 °C min−1. According to the thermogravimetric and thermogravimetric derivative (TG-DTG) curves, two main levels of weight loss are observed in Fig. 2. In the first stage, a weight loss of about 24.5% appeared at the temperature ranging from 171 °C to 220 °C, which is quite similar to the theoretical value 24.3% for releasing two moles of crystal water from MgC2O4·2H2O. In the second stage, another weight loss of about 48.5% could be seen between 407–498 °C analogous to the theoretical value (48.6%) for the decomposition of MgC2O4 by the formation of MgO. These results suggest that during the thermal treatment of MgC2O4·2H2O, it could be completely transformed into MgO at the temperature around 500 °C.
 |
| | Fig. 2 TG-DTG curves of the obtained product by pouring Na2C2O4 into Mg(NO3)2 solution at 50 °C followed by maintaining for 1 h. | |
3.3 TEM observation of the structural transformation
From the in situ TEM observation (Fig. 1), it can be seen that during the decomposition of MgC2O4·2H2O, the surface structure of the resulting product took place a great change along with the variation of crystal structure. To examine the influence of calcination temperature on the surface structure of the resulting MgO particles in the general air condition, we systematically investigated the evolution of the surface structure of the calcined products by TEM. Because the thermal decomposition of MgC2O4·2H2O to MgO mainly occurs in the temperature ranging from 407 °C to 498 °C (Fig. 2), the products by calcination at the temperatures of 400–1000 °C were prepared in a conventional muffle furnace. As shown in Fig. 3, the surface structure along with the crystallization significantly varied with calcination temperature. When the temperature was in the range of 400–600 °C, it is difficult to see through the individual particles (Fig. 3a–c), in good agreement with the results from the in situ TEM observation (Fig. 1a–c). This result illustrates that the temperature range has little effect on the surface structure of the obtained products, which can be confirmed by the HRTEM images. As shown in the left top insets, the obtained products are polycrystalline but consist of domains of single crystalline nanoparticles as shown in the HRTEM images (see Fig. S1 in the ESI†). According to our recent study,15 these particles have the characteristic of mesocrystal materials.39–46 The d spacing between two consecutive lattice planes for the mesocrystals is found to be about 0.21 nm suggesting preferential growth of thermodynamically plane (200) plane. The right top insets give a SAED patterns with the three rings indexed to the (200), (220) and (222) diffractions, respectively, supporting the presence of MgO in cubic phase. From the SAED patterns, it is interesting to be observed that the brightness of the diffraction rings is not uniform, and some spots are very dominant, which could be ascribed to the characteristic of single crystalline as described above. Further evidence could be demonstrated by the HRTEM dark-field images (Fig. S1†), in which it also could be seen that the size of the single crystallite increases with the calcination temperature. This result reveals that the treatment temperature has a pronounced effect on the growth of the MgO crystallite.
 |
| | Fig. 3 (a)–(g) TEM images of the products with variation of calcination temperatures from 400 °C to 1000 °C (top left insets are the corresponding HRTEM images, and top right insets are the SAED patterns). | |
Further increasing the calcination temperature up to 700 °C makes the surface structure of an individual particle clearly observable (Fig. 3d), attributable to the further aggregation or growth of small crystallites. Due to this reason, the d spacing between two consecutive lattice planes decreased to around 0.205 nm (left top inset). Also the SAED pattern (right top inset) demonstrates much difference from those obtained at the temperatures of 400–600 °C, and the diffraction rings are hardly observable just leaving some irregular bright spots, probably resulting from the production of polycrystalline structure (see Fig. S2 in the ESI†). When the temperature was further increased to 800–1000 °C, the surface structure of the typical particle is similar but the size of the crystallite steadily increased from 110 nm to 250 nm (Fig. 3e–g). The d spacing from HRTEM image (left top inset) demonstrates a first increasing trend followed by decreasing. Notablely, all SAED patterns (right top insets) became regular spot matrix, attributable to the production of single crystallites. More interesting is that the SAED pattern gradually changes from monoclinal to cubic structures. It is well-known that MgO only has a cubic structure, and the obtained product in the present study is no exception as confirmed by the XRD patterns as below. From the point of our view, this case might be attributable to the fact as described in Fig. 4. Upon calcining MgC2O4·2H2O at a low temperature, it will release crystal water followed by carbon dioxide and carbon monoxide (Fig. 2) without change in volume, and a pesudomorph similar to the original MgC2O4·2H2O remains. As reported by Eubank, the burning by accompanying the loss of gases would leave considerable space (porosity) between the resulting MgO cells although the original external dimensions of the plane are the same (Fig. 3d–f).22 When the calcination temperature exceeds a certain temperature (e.g., 1000 °C), sintering would take place and the obtained material became denser. Along with the process, recrystallization would become significant and particles grew in size. As shown in Fig. 3g, it is obvious that the crystal size from the calcination temperature of 1000 °C is much bigger than those from 800 °C and 900 °C (Fig. 3e and f). Owing to this fact, the crystalline phase transformed from pesudomorphous (e.g., monoclinal) to cubic structures.
 |
| | Fig. 4 Schematic diagram on the evolution of the crystal plane of MgO from pseudomorphic structure produced from a low temperature calcination (e.g., 800–900 °C) to cubic structure from a high temperature calcination (e.g., 1000 °C). | |
3.4 SEM observation of the surface structure evolution with temperature
To further investigate the evolution of the surface structure of MgO with calcination temperature, the obtained particles were visualized with SEM. For the product (namely MgC2O4·2H2O) by reaction between Na2C2O4 and Mg(NO3)2 at 50 °C followed by drying at 70 °C for 3 h, it demonstrates a fine and smooth surface structure (Fig. 5a). After treating the particles at high temperatures (e.g., 400–600 °C), some fractures appeared at the surface of the particles (Fig. 5b–d), which could be ascribed to the consequence of the releases of crystal water, carbon dioxide and carbon monoxide from the structure of MgC2O4·2H2O. Further increasing the calcination temperature leads to a great change in the surface structures of the resulting products. For example, when the temperature was 700 °C, the surface structure of the particle is composed of nanoparticles with sizes around 80 nm (Fig. 5e). With the increase of the temperature from 800 °C to 1000 °C, the nanoparticles at the surfaces gradually grew from about 88 nm to 120 nm. As reported in the literature,22,26,47–53 the growth in sizes was originated from the sintering of the MgO particles. Although the surface structures of the obtained products significantly changed with calcination temperatures, the external dimensions of the particles almost maintained constant (right top insets of Fig. 5). These results illustrate that for a uniform micro-sized particle such as trapezoid-like MgC2O4·2H2O, the calcination temperature only has a profound effect on its surface structure, and little change was observed in its external contour with temperature.
 |
| | Fig. 5 Typical SEM images of (a) the product by reaction between Na2C2O4 and Mg(NO3)2 and its calcined products at the temperatures of (b) 400 °C, (c) 500 °C, (d) 600 °C, (e) 700 °C, (f) 800 °C, (g) 900 °C, and (h) 1000 °C. Insets are the typical particles in the corresponding calcination temperatures. | |
3.5 XRD patterns of the calcined powders
Fig. 6 shows the X-ray diffraction (XRD) patterns of the baked or calcined powders at 70 °C, 400 °C, 500 °C, 600 °C, 700 °C, 800 °C, 900 °C, and 1000 °C for 3 h. For the product baked at 70 °C, its XRD pattern can be indexed as magnesium oxalate dihydrate (MgC2O4·2H2O, JCPDS no. 28-625). After calcined at temperatures more than 400 °C, the diffraction peak positions and relative intensities for all the samples are in good agreement with cubic MgO (JCPDS no. 4-829), and no other impurities were detected. Careful observation can be found that with increase in the calcination temperatures, the width of the XRD lines becomes narrower, and the intensity of the diffraction peaks steadily gets more and more abundant due to the improved crystallinity which in turn resulted in crystallite growth as described above. Notably, the product calcined at 400 °C also has the characteristic of MgO, but from the TG-DTG curves (Fig. 2) it is obvious that the complete decomposition temperature from MgC2O4·2H2O to MgO is at around 500 °C. The phenomenon might originate from the special structure of MgC2O4 or others, and further investigation is still needed.
 |
| | Fig. 6 XRD patterns of the baked or calcined products at different temperatures as indicated. | |
According to the above discussion, the calcination temperature has a great effect on the sintering of the obtained particles. To quantitatively evaluate the strain and average crystallite size with temperature, a derived Scherrer equation was used.26–28,54
| |
 | (1) |
where
β is the full-width at half-maximum of peak,
θ is the Bragg angle,
Ks is a constant (namely 0.9) and is termed a shape factor,
λ is the wavelength of radiation (0.154056 nm),
D is the relevant crystallite dimension, and
η is the strain associated with the nanoparticles.
Eqn (1) represents a straight line between sin
θ (
X-axis) and
βcos
θ (
Y-axis). The slope of the line gives the strain (
η), and intercept (
Ks ×
λ/
D) of this line on the
Y-axis gives the grain size (
D).
Fig. 7a shows the relation between
βcos
θ and sin
θ for the calcined MgO samples. The straight lines of the calcined MgO at temperatures of 400 °C and 500 °C were almost horizontal, suggesting a lack of strain.
26–28 With the increase in the calcination temperature from 600 °C to 900 °C, the strain of the obtained samples gradually increased except there was a little variation for the sample from 1000 °C. Derived from the slopes and intercepts, the strain and crystallite size of the samples are given in
Fig. 7b. Obviously, with the increase in the temperature, both the obtained strains and crystallite sizes have a similar change pattern. Namely, in the temperature range from 400 °C to 800 °C, both demonstrate an increasing trend responsible for the growth of the calcined samples, followed by a tiny decreasing in the range of 900–1000 °C probably resulting from the shrinkage of the particles due to sintering.
26 Although the
d-spacings from the HRTEM images of the obtained MgO particles also varied with the calcination temperatures (
Fig. 3), no direct correlation was observed between them and the strain values, suggesting that derived Scherrer
eqn (1) is an effective way to evaluate the strain resulting from the calcination. By the way, it is obvious that the calculated crystallite sizes in
Fig. 7b are smaller than those from the measured values from SEM images in
Fig. 5e–h, detailed reasons might be ascribed to the limited range of Scherrer equation in calculation of crystallite size (1–100 nm), or the generation of much error by considering the nanoparticles incorporated at the surface of the micro-sized MgO as monodisperse particles to calculate the crystallite size. In general, the surface structure of a material is generally closely related with its chemical composition. To get an insight into the element components of oxygen (O) atom and magnesium (Mg) atom with temperature, an EDS analysis was carried out for some individual particles from different calcination temperatures.
Fig. 7c presents the plot of the contents of O and Mg atoms from the EDS measurement with varying the temperature. From this figure, it can be seen that as a whole the contents of both O and Mg atoms demonstrate an opposite trend. For example, the content of O atom illustrates a decreasing trend in the temperature range from 400 °C to 800 °C, followed by maintaining in a constant value (
ca. 41%) at the temperatures of 800–1000 °C. However, for Mg atom, it demonstrates a progressively increasing trend at 400–800 °C, and then keeps a value of around 40% at 800–1000 °C, close to the content of O atom. The opposite trend in the range of 400–800 °C could be ascribed to the fact that in the calcination process, O atom will become gas and release from the surface of the samples as the forms of H
2O, CO
2 and CO due to the decomposition of MgC
2O
4·2H
2O (
Fig. 2) and loss of free hydroxyl groups at the surface of MgO, whereas Mg atom is solidified into the form of MgO. At 800–1000 °C, the close values for both O and Mg atoms reveal that in the temperature range, there are trace amounts of crystal or adsorbed H
2O, CO
2 and CO, and the obtained products are almost pure MgO. Due to this, in this range the calcination temperature would have a significant influence on the crystal structure of the resulting MgO as the above mentioned (
Fig. 3).
 |
| | Fig. 7 (a) Plots of βcosθ vs. sinθ for the calcined MgO samples at different temperatures as indicated; (b) changes of strain and crystallite size with calcination temperatures; (c) plots of the contents of oxygen atom and magnesium atom from EDS measurement with variation of calcination temperatures. For the EDS analysis, the atomic component was collected from an individual particle, and each analysis was carried out on three single particles. The average of the obtained data was employed to derive the respective atomic content. Because this study focuses on MgO, the content of carbon atom involved in the component was not included. | |
3.6 Electrochemical properties of the calcined products
In the recent years, MgO as an electrode material has demonstrated its promise in detection of different analytes.7,8,55–57 However, the influence of calcination temperature on the electrochemical performance of MgO is still rare. In this study, we used cyclic voltammetry to explore the electrochemical properties of MgO calcined at different temperatures. Fig. 8a shows the cyclic voltammograms of 1 mM K3Fe(CN)6 in 0.1 M KCl solution (pH = 6.35) measured by using bare carbon paste electrode (CPE) and various MgO modified CPEs. A weak peak current was obtained for 1 mM K3Fe(CN)6 solution at the bare CPE (grey line). Under the same conditions, the peak currents were greatly enhanced when the MgO modified CPEs were employed. For example, the peak current from the MgO calcined at 400 °C (red line) was improved around 2.4-fold relative to the value from the bare CPE. With the increase in the calcination temperature, the cyclic voltammograms varied with the obtained MgO electrodes. Fig. 8b clearly presents the effect of calcination temperature of MgO on the peak current. Apparently, the peak current steadily increased with the calcination temperature of MgO in the range of 400–800 °C followed by demonstrating a decreasing trend (800–1000 °C). Among them, the MgO calcined at 800 °C presents the optimal performance. From the surface structure evolution of MgO with calcination temperatures as described above (Fig. 3), it can be seen that the electrochemical properties of MgO is closely related with its crystal structure. Namely, with the increase of calcination temperature from 400 °C to 1000 °C, the crystalline structure of MgO transformed from mesocrystal (400–600 °C, the mixture of amorphous structure and single crystal) to polycrystal (700 °C), and then to single crystal (800–1000 °C), whereas their electrochemical performances demonstrated a first increasing trend followed by decreasing. This reveals that a more perfect crystal structure of MgO would produce a better electrochemical performance, but a pretty higher calcination temperature (e.g., 900–1000 °C) would make its performance worsen probably attributable to the sintering and shrinkage of MgO. By the way, we also found the content of MgO in CP played a significant role in determining the performance of the modified electrode. As shown in Fig. 8c, with increasing the content of MgO calcined at 800 °C in CP from 0% to 5.5%, the peak current presents an increasing trend, but further increasing the value from 5.5% to 7.5% would make the electrochemical performance sharply decrease. The latter may originate from their higher resistance.58 As well know, MgO is an insulator, and its higher content in CP would conduce to an increase in the resistance of the modified electrodes, thus leading to a decrease in the peak currents.
 |
| | Fig. 8 (a) Cyclic voltammograms of bare carbon paste electrode (CPE) and MgO modified CPEs, and MgO samples were calcined at different temperatures (400–1000 °C); (b) effect of calcination temperature of MgO on the peak current; (c) effect of the content of MgO calcined at 800 °C in carbon paste on the peak current (note: for all the measurements, the sample solution was the mixture of 1 mM K3Fe(CN)6 and 0.1 M KCl, and for (a) and (b), the content of MgO in carbon paste was 5.5% (w/w)). | |
3.7 Catalytic properties of the calcined products
To survey the influence of calcination temperature on the catalytic performance of MgO, the MPV reaction between benzaldehyde and ethanol was carried out. During this reaction, benzaldehyde is reduced to benzyl alcohol while ethanol is oxidized to acetaldehyde. Fig. 9a presents the benzaldehyde conversion efficiency by using the calcined MgO at different temperatures as catalysts for 12 h. It is apparent that the conversion efficiency of benzaldehyde decreased over the calcination temperature, in good agreement with many previous studies59–67 (Table S1 in the ESI†). Specially, although both the calcined samples at 400 °C and 500 °C demonstrated a similar catalytic performance for benzaldehyde (ca. 90% conversion efficiency), a sharp decrease occurred when the calcination temperature was increased to 600 °C followed by slowly decreasing with temperature. From the above discussion, it is obvious that the catalytic activities of the calcined MgO samples are just opposite with their crystalline structure (Fig. 3 and 6), and a poorer quality crystal structure of MgO will lead to a higher catalytic capability. As evidenced by the textural properties of the samples (see Fig. S3 and Table S2 in the ESI†), their catalytic activities are in direct proportion to the surface area and pore volume of MgO, which has a substantial effect on the surface density of the catalyst and influences the reaction kinetics and mass transfer, and hence the overall performance.68
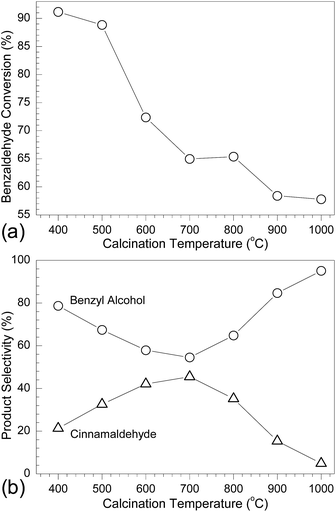 |
| | Fig. 9 Plots of (a) benzaldehyde conversion and (b) product selectivity in the MPV reaction between benzaldehyde and ethanol at 351 K by using the calcined MgO at different temperatures as catalysts. | |
Along with the conversion of benzaldehyde into benzyl alcohol, another side-reaction between benzaldehyde and acetaldehyde from the oxidation of ethanol by production of cinnamaldehyde14,15 also occurred. The present study compares the reaction selectivity for benzyl alcohol and cinnamaldehyde by using the different calcined MgO as catalysts. As shown in Fig. 9b, it is interesting that the reaction selectivity for both products demonstrates a converse correlation with the calcination temperature of MgO. Namely, the selectivity for benzyl alcohol presents a first decreasing pattern followed by increasing with temperature, and it value was the lowest when the temperature was 700 °C. However, cinnamaldehyde appeared a just converse trend, and the selectivity was the highest at 700 °C. These results reveal that in the MPV reaction between benzaldehyde and ethanol, control over the calcination temperature of MgO at a lower or higher value would enable a higher selectivity of benzyl alcohol, whereas the temperature of 700 °C favored the production of cinnamaldehyde with the highest selectivity although the detailed catalytic mechanism is still not clear.
4 Conclusions
In summary, we systematically investigated the influence of calcination temperature on the surface structure and physiochemical capabilities of MgO. To gain a deep insight into the surface structure evolution of trapezoid-like MgC2O4·2H2O by production of MgO, analyses were carried out by using different characterization techniques including TEM, TGA, SEM and XRD. The calcination temperature was found to be vital in determining the final crystal structure of the resulting MgO. Its structure evolved from mesocrystal in the temperature range of 400–600 °C to polycrystallite at 700 °C, then to pesudomorphous MgO at 800–900 °C, and finally to cubic single crystal at an elevated temperature 1000 °C. Along with the process, its surface changed from smooth to fractured structures followed by the structure composed of uniform nanoparticles resulting from the sintering of MgO at a higher temperature. These surface properties (e.g., crystalline structure) were found to be responsible for the electrochemical properties of MgO, whereas their catalytic activities were highly dependent on the textural properties (e.g., surface area and porosity) although the reaction selectivity was still closely related with calcination temperature. We believe that this knowledge not only greatly promotes our better understanding on the process of the surface structure evolution of MgO during calcination at a high temperature, but also provides a deep insight into the correlation between its physicochemical properties and functions.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21205093), Key Science and Technology Program of Shaanxi Province of China (No. 2014K13-16), Special Research Project of Shaanxi Provincial Department of Education in China (2013JK0674), Xi'an Science and Technology Project in China (No. CXY1434(6)) and Graduate Innovation Foundation of Xi'an Shiyou University (No. 2014cx130737).
Notes and references
- J. Guzman and B. C. Gates, Angew. Chem., Int. Ed., 2003, 42, 690–693 CrossRef CAS PubMed.
- M. F. Hoq and K. J. Klabunde, J. Am. Chem. Soc., 1986, 108, 2114–2116 CrossRef CAS.
- P. Schwach, M. G. Willinger, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 11381–11384 CrossRef CAS PubMed.
- C.-Y. Cao, J. Qu, F. Wei, H. Liu and W.-G. Song, ACS Appl. Mater. Interfaces, 2012, 4, 4283–4287 CAS.
- Y.-X. Li, H. Li and K. J. Klabunde, Environ. Sci. Technol., 1994, 28, 1248–1253 CrossRef CAS PubMed.
- Z. Zhang, Y. Zheng, J. Chen, Q. Zhang, Y. Ni and X. Liang, Adv. Funct. Mater., 2007, 17, 2447–2454 CrossRef CAS PubMed.
- L. Lu, L. Zhang, X. Zhang, Z. Wu, S. Huan, G. Shen and R. Yu, Electroanalysis, 2010, 22, 471–477 CrossRef CAS PubMed.
- Z. Wu, C. Xu, H. Chen, Y. Wu, H. Yu, Y. Ye and F. Gao, J. Phys. Chem. Solids, 2013, 74, 1032–1038 CrossRef CAS PubMed.
- A. Iovan, S. Andersson, Y. G. Naidyuk, A. Vedyaev, B. Dieny and V. Korenivski, Nano Lett., 2008, 8, 805–809 CrossRef CAS PubMed.
- K. P. McKenna, D. Koller, A. Sternig, N. Siedl, N. Govind, P. V. Sushko and O. Diwald, ACS Nano, 2011, 5, 3003–3009 CrossRef CAS PubMed.
- J. V. Stark and K. J. Klabunde, Chem. Mater., 1996, 8, 1913–1918 CrossRef CAS.
- K. Zhu, J. Hu, C. Kübel and R. Richards, Angew. Chem., Int. Ed., 2006, 45, 7277–7281 CrossRef CAS PubMed.
- N. Sutradhar, A. Sinhamahapatra, S. K. Pahari, P. Pal, H. C. Bajaj, I. Mukhopadhyay and A. B. Panda, J. Phys. Chem. C, 2011, 115, 12308–12316 CAS.
- F. Wang, N. Ta and W. Shen, Appl. Catal., A, 2014, 475, 76–81 CrossRef CAS PubMed.
- X. Zhang, Y. Zheng, H. Yang, Q. Wang and Z. Zhang, CrystEngComm, 2015, 17, 2642–2650 RSC.
- Y. Hao, G. Meng, C. Ye, X. Zhang and L. Zhang, J. Phys. Chem. B, 2005, 109, 11204–11208 CrossRef CAS PubMed.
- Y. Ding, G. Zhang, H. Wu, B. Hai, L. Wang and Y. Qian, Chem. Mater., 2001, 13, 435–440 CrossRef CAS.
- H. Minami, K. Kinoshita, T. Tsuji and H. Yanagimoto, J. Phys. Chem. C, 2012, 116, 14568–14574 CAS.
- B. Eckhardt, E. Ortel, J. Polte, D. Bernsmeier, O. Görke, P. Strasser and R. Kraehnert, Adv. Mater., 2012, 24, 3115–3119 CrossRef CAS PubMed.
- F. Mohandes, F. Davar and M. Salavati-Niasari, J. Phys. Chem. Solids, 2010, 71, 1623–1628 CrossRef CAS PubMed.
- Z. P. Zhang, Y. J. Zheng, Y. W. Ni, Z. M. Liu, J. P. Chen and X. M. Liang, J. Phys. Chem. B, 2006, 110, 12969–12973 CrossRef CAS PubMed.
- W. R. Eubank, J. Am. Ceram. Soc., 1951, 34, 225–229 CrossRef CAS PubMed.
- P. L. Gai, J. M. Montero, A. F. Lee, K. Wilson and E. D. Boyes, Catal. Lett., 2009, 132, 182–188 CrossRef CAS.
- M. S. Mastuli, R. Rusdi, A. M. Mahat, N. Saat and N. Kamarulzaman, Adv. Mater. Res., 2012, 545, 137–142 CrossRef CAS.
- C. M. Pathak and V. K. Moorthy, Trans. Indian Ceram. Soc., 2014, 35, 89–98 CrossRef PubMed.
- T. Ikegami, M. Kobayashi, Y. Moriyoshii, S.-I. Shirasaki and H. Suzuki, J. Am. Ceram. Soc., 1980, 63, 640–643 CrossRef CAS PubMed.
- D. K. Thomas and T. W. Baker, Proc. Phys. Soc., London, 1959, 74, 673 CrossRef CAS.
- P. J. Anderson and D. T. Livey, Powder Metall., 1961, 4, 189–203 CrossRef PubMed.
- E. Matijevic, Langmuir, 1994, 10, 8–16 CrossRef CAS.
- J.-G. Li, X. Li, X. Sun, T. Ikegami and T. Ishigaki, Chem. Mater., 2008, 20, 2274–2281 CrossRef CAS.
- T. Yokoi, J. Wakabayashi, Y. Otsuka, W. Fan, M. Iwama, R. Watanabe, K. Aramaki, A. Shimojima, T. Tatsumi and T. Okubo, Chem. Mater., 2009, 21, 3719–3729 CrossRef CAS.
- C. Fenzl, S. Wilhelm, T. Hirsch and O. S. Wolfbeis, ACS Appl. Mater. Interfaces, 2013, 5, 173–178 CAS.
- J. Flemke, M. Maywald and V. Sieber, Biomacromolecules, 2013, 14, 207–214 CrossRef CAS PubMed.
- R. B. Karyappa and R. M. Thaokar, Langmuir, 2014, 30, 10270–10279 CrossRef CAS PubMed.
- N. Vogel, M. Retsch, C.-A. Fustin, A. del Campo and U. Jonas, Chem. Rev., 2015, 115, 6265–6311 CrossRef CAS PubMed.
- X. Zhang, Y. Zheng, H. Yang, Q. Wang and Z. Zhang, RSC Adv., 2015, 5, 63034–63043 RSC.
- https://en.wikipedia.org/wiki/Electron_beam_technology.
- W. Zhu, L. Zhang, G.-L. Tian, R. Wang, H. Zhang, X. Piao and Q. Zhang, CrystEngComm, 2014, 16, 308–318 RSC.
- T. Li, Z. Cao, H. You, M. Xu, X. Song and J. Fang, Chem. Phys. Lett., 2013, 555, 154–158 CrossRef CAS PubMed.
- Y.-Y. Kim, A. S. Schenk, J. Ihli, A. N. Kulak, N. B. J. Hetherington, C. C. Tang, W. W. Schmahl, E. Griesshaber, G. Hyett and F. C. Meldrum, Nat. Commun., 2014, 5, 4341 CAS.
- X. Liang, L. Gao, S. Yang and J. Sun, Adv. Mater., 2009, 21, 2068–2071 CrossRef CAS PubMed.
- J. Fang, B. Ding, X. Song and Y. Han, Appl. Phys. Lett., 2008, 92, 173120 CrossRef PubMed.
- H. You, Y. Ji, L. Wang, S. Yang, Z. Yang, J. Fang, X. Song and B. Ding, J. Mater. Chem., 2012, 22, 1998 RSC.
- S.-J. Liu, J.-Y. Gong, B. Hu and S.-H. Yu, Cryst. Growth Des., 2008, 9, 203–209 Search PubMed.
- J. Fang, B. Ding and H. Gleiter, Chem. Soc. Rev., 2011, 40, 5347–5360 RSC.
- C. You, Q. Zhang, Q. Jiao and Z. Fu, Cryst. Growth Des., 2009, 9, 4720–4724 CAS.
- H. C. Fischer, J. Am. Ceram. Soc., 1955, 38, 245–251 CrossRef CAS PubMed.
- J.-G. Yu, H.-G. Yu, B. Cheng, X.-J. Zhao, J. C. Yu and W.-K. Ho, J. Phys. Chem. B, 2003, 107, 13871–13879 CrossRef CAS.
- Y. X. Pang and X. Bao, J. Eur. Ceram. Soc., 2003, 23, 1697–1704 CrossRef CAS.
- G. Lusvardi, G. Malavasi, V. Aina, L. Bertinetti, G. Cerrato, G. Magnacca, C. Morterra and L. Menabue, Langmuir, 2010, 26, 10303–10314 CrossRef CAS PubMed.
- N. Xiao, Z. Li, J. Liu and Y. Gao, Thin Solid Films, 2010, 519, 541–548 CrossRef CAS PubMed.
- R. H. Krishna, B. M. Nagabhushana, H. Nagabhushana, N. S. Murthy, S. C. Sharma, C. Shivakumara and R. P. S. Chakradhar, J. Phys. Chem. C, 2013, 117, 1915–1924 CAS.
- A. Worawong, T. Jutarosaga and W. Onreabroy, Adv. Mater. Res., 2014, 979, 208–211 CrossRef CAS.
- G. K. Williamson and W. H. Hall, Acta Metall., 1953, 1, 22–31 CrossRef CAS.
- A. Umar, M. M. Rahman and Y.-B. Hahn, Electrochem. Commun., 2009, 11, 1353–1357 CrossRef CAS PubMed.
- Y.-Y. Ling, Q.-A. Huang, D.-X. Feng, X.-Z. Li and Y. Wei, Anal. Methods, 2013, 5, 4580–4584 RSC.
- Y. Wei, R. Yang, X.-Y. Yu, L. Wang, J.-H. Liu and X.-J. Huang, Analyst, 2012, 137, 2183–2191 RSC.
- G. B. Soares, C. M. P. Vaz, C. Ribeiro and I. Hermans, Electrocatalysis, 2014, 6, 92–101 CrossRef.
- F. Li, H. Li, L. Wang, P. He and Y. Cao, Catal. Sci. Tech., 2015, 5, 1021–1034 RSC.
- J. Li, Y. le, W.-L. Dai, H. Li and K. Fan, Catal. Commun., 2008, 9, 1334–1341 CrossRef CAS PubMed.
- J. K. Bartley, C. Xu, R. Lloyd, D. I. Enache, D. W. Knight and G. J. Hutchings, Appl. Catal., B, 2012, 128, 31–38 CrossRef CAS PubMed.
- L. Wang, H. Li, S. Xin and F. Li, Catal. Commun., 2014, 50, 49–53 CrossRef CAS PubMed.
- M. Kong, Q. Yang, W. Lu, Z. Fan, J. Fei, X. Zheng and T. D. Wheelock, Chin. J. Catal., 2012, 33, 1508–1516 CrossRef CAS.
- K. M. Mothi, G. Soumya and S. Sugunan, Bull. Chem. React. Eng. Catal., 2014, 9, 175–181 CAS.
- P. L. Tan, Y. L. Leung, S. Y. Lai and C. T. Au, Appl. Catal., A, 2002, 228, 115–125 CrossRef CAS.
- Z. Zhang, Q. Zhu, J. Ding, X. Liu and W.-L. Dai, Appl. Catal., A, 2014, 482, 171–178 CrossRef CAS PubMed.
- J. He, G.-Q. Xie, J.-Q. Lu, L. Qian, X.-L. Zhang, P. Fang, Z.-Y. Pu and M.-F. Luo, J. Catal., 2008, 253, 1–10 CrossRef CAS PubMed.
- H. Tüysüz and F. Schüth, in Advances in Catalysis, ed. C. G. Bruce and C. J. Friederike, Academic Press, 2012, vol. 55, pp. 127–239 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HRTEM images of the calcined MgO at the temperatures of 400 °C, 500 °C and 600 °C and 700 °C (Fig. S1); N2 adsorption–desorption isotherms and BJH pore size distribution curves of the calcined MgO samples at different temperatures ranging from 400 °C to 1000 °C (Fig. S2); effect of calcination temperature on catalytic performance (Table S1); texture properties of as-synthesized MgO calcined at different temperatures (Table S2). See DOI: 10.1039/c5ra17031a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 









