
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self assembled nanocages from DNA–protoporphyrin hybrid molecules†
Vandana
Singh
a,
Mohan
Monisha
b,
Roy
Anindya
b and
Prolay
Das
*a
aDepartment of Chemistry, Indian Institute of Technology Patna, Patna-800013, Bihar, India. E-mail: prolay@iitp.ac.in; Fax: +91 612 225 7383; Tel: +91 612 255 2057
bIndian Institute of Technology Hyderabad, Hyderabad-502205, India
First published on 15th October 2015
Abstract
DNA–organic hybrid molecular building blocks are generated by covalent conjugation of the carboxyl groups of protoporphyrin IX with the amine functional groups of modified DNA oligomers. The hybrids are used to engineer DNA nanocages by self-assembly of the complementary DNA–organic molecule conjugates. The nanocages were found to be lined up in head to tail fashion under the selective ionic strength of the solution. Computational approach revealed the area and volume acquired by each DNA–organic hybrid nanocages.
The current trends in structural DNA nanotechnology are to create predictable DNA nanostructures that involve DNA–organic hybrid molecules.1 These hybrid molecular building block based nanostructures find potential applications as probes, for creation of proteins arrays, molecular electronics, single nucleotide polymorphism, RNAomics, drug encapsulation and delivery, light harvesting, bio sensing and others.2–10 DNA–organic hybrids have the added advantages of better base pair fidelity, stability, directionality and DNA economy.11–13 However, apart from offering stability and flexible directionality, the electronic and physical properties of the organic molecule in the DNA–organic hybrids often remains unexplored. DNA organic hybrid nanostructures have the potential to harbour a combination drug. Such combinations may be created by using two different organic molecules to be linked to DNA in the nanostructures. Cage like DNA-nanostructures has also been shown to encapsulate various proteins.14,15 Herein, we report the facile creation of nanocages from DNA–protoporphyrin IX (PpIX) hybrid molecular building blocks through self-assembly and explore their applications that involve the self-assembled nanocages as well as the organic molecule PpIX.
PpIX has been explored explicitly for potential applications in photodynamic therapy (PDT).16–18 There are certain advantages of using PpIX as the organic counterpart in DNA–organic hybrid molecules as it is biomimetic in origin.19 PpIX is a π-electron rich light sensitive molecule and thus envisioned to have multiple applications involving conductivity, FRET, ROS and sensing volatile organic compounds (VOCs).20–24 Recently, cationic porphyrin–tetrapeptide conjugates were synthesized which are good candidates for peptide delivery.25 Free base porphyrin acting as molecular glue is used to assemble non-complementary DNA sequences at ionic strength greater than 60 mM.26 Although, porphyrin has been conjugated with DNA to create various nanoassemblies, PpIX, an important member of the porphyrin family has not featured in any of the nanostructures that involve a porphyrin compound. As such, there is no report of DNA–PpIX based creation of distinct nanostructures. For the first time, we report the formation of definite nanostructures in the form of cages from the hybridization based self-assembly of complementary DNA that is covalently conjugated to PpIX (Scheme 1).
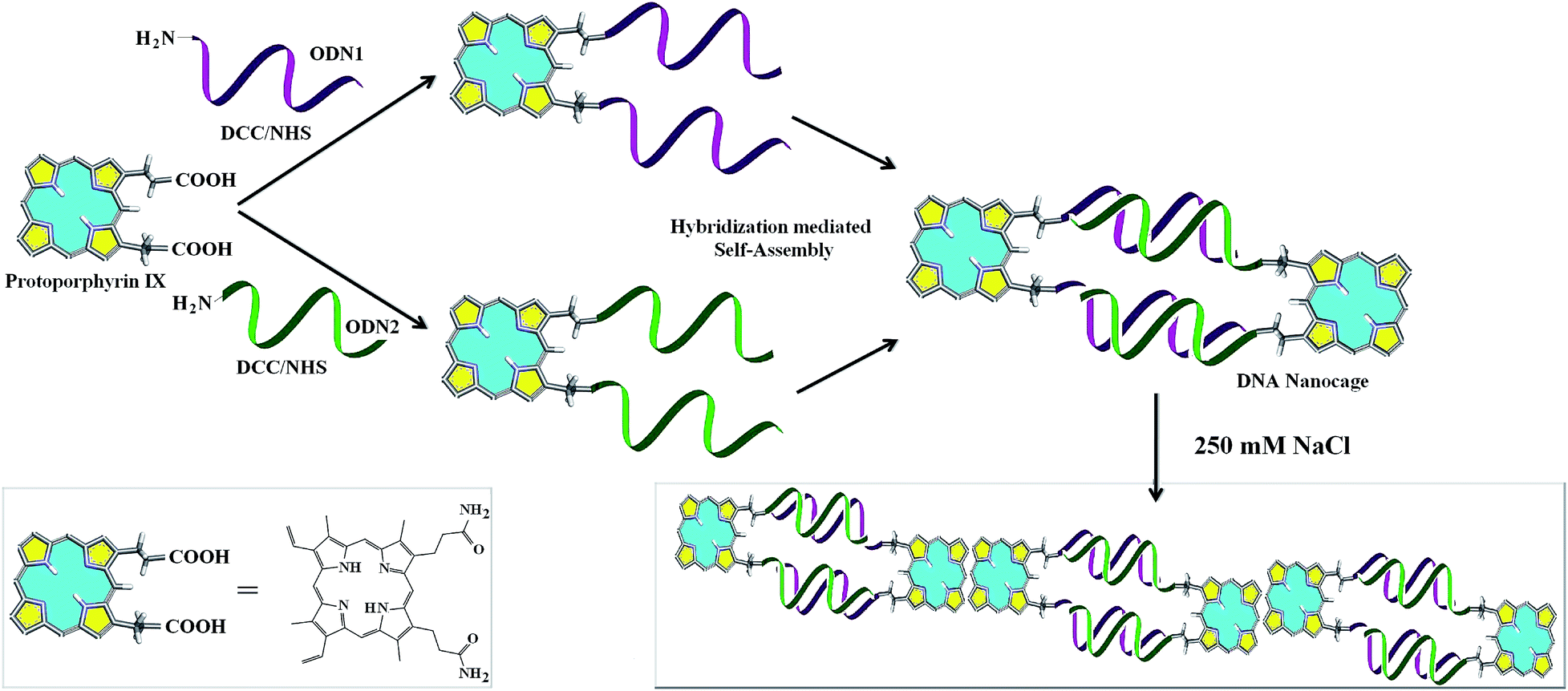 | ||
| Scheme 1 Creation of nanocages from DNA–PpIX hybrid conjugates by self-assembly. | ||
The DCC/NHS mediated chemistry was used to couple the carboxyl groups of PpIX separately with the 5′-amine terminated 12 bases long single strand DNA (ssDNA, ODN1) and its complementary strand (ODN2). Due to the presence of two carboxyl groups in PpIX, two reaction products were formed depending on the number of ssDNA that are coupled to a single PpIX molecule (Fig. 1). The basicity of the medium favours the conjugation of oligonucleotides with PpIX, which is reflected in a higher yield of (ODN1)2–PpIX and (ODN2)2–PpIX by ∼33% at pH 8 than at pH 7. The monoconjugated and the diconjugated ODN–PpIX species are well separated in denaturing PAGE (Fig. 1, ESI 1 and 2†). The upper bands in the denaturing PAGE corresponds to the diconjugated (ODN1)2–PpIX and (ODN2)2–PpIX, while the lower bands correspond to ODN1–PpIX, ODN2–PpIX and unreacted ODNs. The (ODN1)2–PpIX and (ODN2)2–PpIX were extracted from the gel and purified for auxiliary characterizations and subsequent self-assembly experiments.
 | ||
| Fig. 1 25% denaturing PAGE showing successful conjugation of PpIX with ODN1 and ODN2. | ||
The resulting two DNA–PpIX conjugates formed were further confirmed by HPLC and MALDI-ToF Mass Spectrometry (MS) (ESI 3–5 and 6–9†). Through MALDI-ToF mass spectrometry, it was observed that both the single conjugated and diconjugated oligonucleotides–PpIX conjugates are formed. The % intensity of diconjugates, (ODN1)2–PpIX and (ODN2)2–PpIX was more as compared to the single conjugated ODN1–PpIX and ODN2–PpIX (ESI 3†). The peaks that were observed in HPLC corresponds to the unreacted DNA, single and di-conjugated DNA–PpIX and unreacted PpIX (ESI 6†). With covalent attachment of a hydrophobic molecule like PpIX to ssDNA, the retention time increases than normal DNA, which is even more for the species that contains two ssDNA attached to a single PpIX molecule. Higher peak intensity for the di-conjugates further confirms preferable formation of the hybrid under the given reaction conditions.27
The diconjugated DNA–PpIX hybrids (ODN1)2–PpIX and (ODN2)2–PpIX were hybridized in the presence of sodium chloride that lead to the self-assembly of the DNA–PpIX hybrids (ESI†). Solution mixture of (ODN1)2–PpIX (1 nmol) and (ODN2)2–PpIX (1 nmol) was heated to 90 °C and then slowly cooled to 20 °C with a ramp of 0.1°C s−1 in the presence of 10 mM TE, 10 mM magnesium chloride and 250 mM NaCl. This hybridization lead to the formation of higher ordered structures that are characterized indirectly and directly by various analytical techniques. Native PAGE shows the formation of DNA bands corresponding to the dimeric form of ODN1–ODN2 (12 × 2 × 2 = 48 bases). However, smeared bands with low gel mobility was also observed for the self-assembled (ODN1)2–PpIX and (ODN2)2–PpIX. These bands indicate that higher order structures are prominently formed at the hybridization condition used (Fig. 2). The position of the bands with respect to the ladder and discreteness suggest that self-assembly lead to the distribution of higher ordered structures where four or more dimeric units (equivalent to ∼200 bp and more) may be aligned together. Occurrence of higher ordered structures upon self-assembly of hybrids was further confirmed by dynamic light scattering studies (DLS). The DNA duplex has a pitch of 0.33 nm per base pair and a diameter of 2 nm. However, cumulant apparent hydrodynamic radius of around ∼54 nm was recorded following hybridization of (ODN1)2–PpIX and (ODN2)2–PpIX (ESI 10 and 11†). The particle size inferred from DLS, points towards the aggregation of self-assembled DNA hybrids.
 | ||
| Fig. 2 10% native PAGE gel showing formation of higher ordered structure from self-assembly (ODN1)2–PpIX–(ODN2)2–PpIX in 250 mM NaCl and 10 mM MgCl2 and 10 mM TE. | ||
Thermal melting temperature (Tm) of self-assembled (ODN1)2–PpIX and (ODN2)2–PpIX increased by 3 °C as compared to the Tm of ODN1–ODN2, which is 54 °C (Fig. 3). The increase in thermal melting of self-assembled hybrids are attributed to the reduced configurational entropy and ion cloud sharing of the surrounding duplex DNA, coupled with increasing sticky end associations due to self-assembly.13,28,29 The melting temperature of ODN1–ODN2 remains unchanged in the presence of free PpIX molecule at concentration equivalent to that in the DNA–PpIX hybrids. This indicates that an increase in melting temperature is not due to intercalation of PpIX on the DNA.
 | ||
| Fig. 3 Thermal melting profile of normal ODN1–ODN2 and (ODN1)2–PpIX–(ODN2)2–PpIX. | ||
The direct evidence of nano-assemblies formation was given by AFM studies (Fig. 4). Cage like morphology was observed for the self-assembled (ODN1)2–PpIX–(ODN2)2–PpIX. Each cage like structure formed is of ∼10 nm pertaining to head to tail distances of suffixed PpIX at both the ends of DNA nanocages. Interestingly, these dispersed 2D cages are also found to be aggregated in head to tail fashion to form staircase case like ordered structures. This phenomenon is not uncommon for pi-electron rich porphyrin type molecules. Such molecules are inclined to interact with neighbouring similar molecules at elevated salt concentration.30 This also explains the higher order bands in the native PAGE (Fig. 2). A wide distribution of these aggregates results in multiple bands with limited mobility in the PAGE.
 | ||
| Fig. 4 AFM image showing self assembled (ODN1)2–PpIX–(ODN2)2–PpIX leading to the formation explicit 2D nanocages. | ||
The CD spectrum of the self-assembled DNA–PpIX conjugates in the presence of 250 mM of NaCl were markedly different from the normal hybridized oligonucleotide duplex (Fig. 5). The changes in the positive and negative band at 280 and 245 nm respectively, for the self-assembled structures indicate a conformational constraint in the DNA duplex. (DNA)2–PpIX conjugates tend to aggregate due to π–π stacking of the aromatic PpIX.31 The significant changes in the characteristic peak of DNA below 250 nm is attributed to the head to tail attachment of DNA–PpIX conjugate and loss of helicity upon cage formation of extended structures. Decrease in molar positive (∼31%) and negative ellipticity (∼68%) of (ODN1)2–PpIX–(ODN2)2–PpIX suggests deformation in the native B form of the DNA.
 | ||
| Fig. 5 Circular dichroism of normal ODN1–ODN2 and (ODN1)2–PpIX–(ODN2)2–PpIX. | ||
The emission spectra of self-assembled (ODN1)2–PpIX and (ODN2)2–PpIX are distinctly different from the diconjugated hybrids (Fig. 6). At excitation wavelength of 400 nm, there is a considerable decrease in the intensity of the signature emission peaks of (ODN1)2–PpIX at 627 nm and 691 nm in (ODN1)2–PpIX–(ODN2)2–PpIX at 250 mM concentration of salt. The bands with low intensity is attributed to the residual non-stacked PpIX at two ends of each system.32 As the concentration of salt is increased to 350 mM, more of (ODN1)2–PpIX and (ODN2)2–PpIX aggregate together in head to tail fashion at 25 °C. This results in further decrease in intensity of emission of PpIX. However, the unaltered peak position of PpIX in the emission spectrum of the self-assembly indicate that the photo-physical properties of PpIX is retained in the nanostructure. The emission spectra further confirm that closed loop structure formation does not take place upon hybridization, that would alter the spectra significantly. Such structures were also not observed in AFM studies since the probability of their formation is entropically and enthalpically disfavoured.32
 | ||
| Fig. 6 Emission spectra of (ODN1)2–PpIX–(ODN2)2–PpIX (λexc = 400 nm). Number of nanocages depicted are symbolic to denote more aggregation at higher salt concentration. | ||
Computational methods were engaged to theoretically evaluate the dimension of the nanocages formed from self assembly of (ODN1)2–PpIX and (ODN2)2–PpIX. Each of these protoporphyrin IX conjugated ssDNA structures was associated to constitute the DNA nanocages, where each of the di-conjugated DNA–PpIX units are held together by the hydrophobic interactions. The total length of a single DNA nanocage was found to be approximately 9.6 nm. This is in excellent agreement with the experimentally determined length from AFM studies. The total surface area and the volume of each DNA nanocage unit was found to be 8406 Å2 and 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 640 Å3 respectively (Fig. 7 and ESI 12†). These were calculated using 3 V: cavity, channel and cleft volume calculator and extractor.33,34
640 Å3 respectively (Fig. 7 and ESI 12†). These were calculated using 3 V: cavity, channel and cleft volume calculator and extractor.33,34
 | ||
| Fig. 7 Computational studies depicting explicit nanocages formation. The structure of DNA and PpIX are represented in sticks with carbon, nitrogen, oxygen and phosphate atoms shown in green, blue, red and orange colors respectively using the PyMol molecular graphics system with head to tail length of 9.6 nm. | ||
The use of solution phase chemistry to synthesize DNA–organic molecules has proved to be a simpler alternative to DNA synthesizer based approach. Post-synthetically created DNA–organic hybrids exhibit better directionality and DNA economy while creating a nanostructure. The use of 5′-NH2 terminated oligonucleotides for conjugation with PpIX is a novice effort to be used in molecular recognition. Previously, we have reported the creation of DNA–PpIX conjugate that was used as a FRET acceptor for ROS generation.24 Here, we have demonstrated the creation of distinct nanostructures from self-complementary DNA strands that are covalently attached to PpIX. The conjugates are well characterized and subsequently self-assembled through DNA hybridization. The self-assembly lead to the formation of cage like structures whose length and dimensions are determined by the length of the DNA sequence. Furthermore, such nanocages were found to align themselves in head to tail position forming extended structures resembling a ladder. Hypothetically, a 1-dimensional linear array formation is also a possibility, where two DNA–PpIX hybrids would be held together with a single DNA duplex formation. This would engage the two hybrids in every alternate position and go hand-in-hand to form extended linear arrays. However, such structures were not detected under the given experimental conditions. This indicates towards the significant contribution of the PpIX molecule to influence the outcome of the DNA-self assembly. It is desired that the electronic and photophysical properties of the organic counterpart in DNA–organic hybrids be retained to introduce versatility in the nanostructure for multifaceted applications. The DNA–PpIX nanostructures could find potential applications in biochip formation, coherent ROS generation and also to hold proteins, drug molecules or other nanoparticles. Alternatively, the DNA strands can be wisely chosen to be aptamers for relevant cell surface marker proteins, whereby release of a cargo can be envisaged by the opening of the nanocages due to DNA–protein interaction.
Acknowledgements
Funding from Department of Biotechnology (DBT), Govt. of India (project no. BT/PR3444/NNT/28/560/2011) to P. D is gratefully acknowledged. V. S. is thankful to Central Scientific and Industrial Research (CSIR), Govt. of India (Sanction no.09/1023(0004)/2010-EMR-1). IIT Patna is acknowledged for providing infrastructural and experimental facilities.Notes and references
- N. C. Seeman and A. M. Belcher, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6451–6455 CrossRef CAS PubMed.
- P. M. Dentinger, B. A. Simmons, E. Cruz and M. Sprague, Langmuir, 2006, 22, 2935–2937 CrossRef CAS PubMed.
- W. W. Zhao, J. J. Xu and H. Y. Chen, Chem. Rev., 2014, 114, 7421–7441 CrossRef CAS PubMed.
- H. Pei, X. Zuo, D. Pan, J. Shi, Q. Huang and C. Fan, NPG Asia Mater., 2013, 5, e51 CrossRef CAS PubMed.
- Y. Zhao, H. Wang, W. Tang, S. Hu, N. Li and F. Liu, Chem. Commun., 2015, 51, 10660–10663 RSC.
- Y. Cho, J. B. Lee and J. Hong, Sci. Rep., 2014, 4, 4078 Search PubMed.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS PubMed.
- J. K. Lee, Y. H. Jung, J. B. H. Tok and Z. Bao, ACS Nano, 2011, 5, 2067–2074 CrossRef CAS PubMed.
- C. J. Serpell, T. G. W. Edwardson, P. Chidchob, K. M. M. Carneiro and H. F. Sleiman, J. Am. Chem. Soc., 2014, 136, 15767–15774 CrossRef CAS PubMed.
- C. K. McLaughlin, G. D. Hamblina and H. F. Sleiman, Chem. Soc. Rev., 2011, 40, 5647–5656 RSC.
- Z. Dogan, R. Paulini, J. A. Rojas Stütz, S. Narayanan and C. Richert, J. Am. Chem. Soc., 2004, 126, 4762–4763 CrossRef CAS PubMed.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- I. Yildirim, I. Eryazici, S. T. Nguyen and G. C. Schatz, J. Phys. Chem. B, 2014, 118, 2366–2376 CrossRef CAS PubMed.
- J. D. Flory, C. R. Simmons, S. Lin, T. Johnson, A. Andreoni, J. Zook, G. Ghirlanda, Y. Liu, H. Yan and P. Fromme, J. Am. Chem. Soc., 2014, 136, 8283–8295 CrossRef CAS PubMed.
- R. Crawford, C. M. Erben, J. Perizl, L. M. Hall, T. Brown, A. J. Turberfield and A. N. Kapanidis, Angew. Chem., Int. Ed., 2013, 52, 2284–2288 CrossRef CAS PubMed.
- J. C. Kennedy, R. H. Pottier and D. C. Pross, J. Photochem. Photobiol., B, 1990, 6, 143–148 CrossRef CAS.
- S. Anand, B. J. Ortel, S. P. Pereira, T. Hasan and E. V. Maytin, Cancer Lett., 2012, 326, 8–16 CrossRef CAS PubMed.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- P. Ling, Q. Hao, J. Lei and H. Ju, J. Mater. Chem. B, 2015, 3, 1335–1341 RSC.
- J. G. Levy, Semin. Oncol., 1994, 21, 4–10 CAS.
- B. Diez, R. C. Russo, M. J. Teijo, S. Hajos, A. Batlle and H. Fukuda, Cell. Mol. Biol., 2009, 55, 15–19 CAS.
- G. Sedghi, K. Sawada, L. J. Esdaile, M. Hoffmann, H. L. Anderson, D. Bethell, W. Haiss, S. J. Higgins and R. J. Nichols, J. Am. Chem. Soc., 2008, 130, 8582–8583 CrossRef CAS PubMed.
- J. Roales, J. M. Pedrosa, M. Cano, M. G. Guillen, M. G. T. Lopes-Costa, P. Castillero, A. Barranco and A. R. Gonzalez-Elipe, RSC Adv., 2014, 4, 1974–1981 RSC.
- S. Singh, A. Chakraborty, V. Singh, A. Molla, S. Hussain, M. K. Singh and P. Das, Phys. Chem. Chem. Phys., 2015, 17, 5973–5981 RSC.
- G. Mező, L. Herényi, J. Habdas, Z. Majer, B. Myśliwa-Kurdziel, K. Tóth and G. Csík, Biophys. Chem., 2011, 155, 36–44 CrossRef PubMed.
- G. Sargsyan and M. Balaz, Org. Biomol. Chem., 2012, 10, 5533–5540 CAS.
- M. Endo, T. Royama, M. Fujitsuka and T. Majima, J. Org. Chem., 2005, 70, 7468–7472 CrossRef CAS PubMed.
- J. Nangreave, H. Yan and Y. Liu, Biophys. J., 2009, 97, 563–571 CrossRef CAS PubMed.
- A. A. Greschner, V. Toader and H. F. Sleiman, J. Am. Chem. Soc., 2012, 134, 14382–14389 CrossRef CAS PubMed.
- C. S. Braun, G. S. Jas, S. Choosakoonkriang, G. S. Koe, J. G. Smith and C. R. Middaugh, Biophys. J., 2003, 84, 1114–1123 CrossRef CAS.
- P. P. Neelakandan, Z. Pan, M. Hariharan, Y. Zheng, H. Weissman, B. Rybtchinski and F. D. Lewis, J. Am. Chem. Soc., 2010, 132, 15808–21581 CrossRef CAS PubMed.
- A. Mammana, G. Pescitelli, T. Asakawa, S. Jockusch, A. G. Petrovic, R. R. Monaco, R. Purrello, N. J. Turro, K. Nakanishi, G. A. Ellestad, M. Balaz and N. Berova, Chem.–Eur. J., 2009, 15, 11853–11866 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- N. R. Voss and M. Gerstein, Nucleic Acids Res., 2010, 38, W555–W562 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and data related to HPLC, MALDI ToF, DLS is available here. See DOI: 10.1039/c5ra16851a |
| This journal is © The Royal Society of Chemistry 2015 |