
Ganocochlearic acid A, a rearranged hexanorlanostane triterpenoid, and cytotoxic triterpenoids from the fruiting bodies of Ganoderma cochlear†
Xing-Rong Pengab,
Xia Wangab,
Lin Zhouab,
Bo Houab,
Zhi-Li Zuoa and
Ming-Hua Qiu*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, People's Republic of China. E-mail: mhchiu@mail.kib.ac.cn; Fax: +86-0871-5223325; Tel: +86-0871-5223327
bGraduate University of the Chinese Academy of Sciences, Beijing 100049, People's Republic China
First published on 29th October 2015
Abstract
Ganocochlearic acid A (1), a rearranged hexanorlanostane triterpenoid featuring a γ-lactone ring and a five-membered carbon ring, and eleven new lanostane triterpenoids (2–12), along with six known analogues (13–18) were isolated from the fruiting bodies of Ganoderma cochlear. Their structures, including absolute configurations, were established on the basis of the MS, NMR, X-ray crystallographic, and ECD analysis. A plausible biosynthetic pathway for 1 was proposed. Compounds 7–9, 11–13 showed moderate cytotoxic activities against five human tumor cell lines (HL-60, SMMC-7721, A-549, MCF-7 and SW480) with IC50 values ranging from 8 to 30 μM. Compound 4 exhibited relatively potent cytotoxic activity against MCF-7 cells (IC50: 9.15 μM), compared to the positive control (cisplatin, IC50: 12.7 μM).
Introduction
Ganoderma (Ganodermataceae) is a popular dietary supplement in East Asia, where it is taken to enhance health and longevity. Its use in traditional Chinese and other Asian medicines in the treatment of different diseases, especial various types of cancers, and is backed by two thousand years of anecdotal evidence and a growing body of scientific data.1–5 Lanostane triterpenoids are one of the main groups, which are known to inhibit cancer growth and metastasis by modulating the immune system, inducing cell-cycle arrest, and triggering apoptosis.6–9 Ganoderma lucidum is probably the most well-studied species of all the lanostane-containing fungi, nevertheless, other species of the same genus have also been studied and identified as good sources of potential anticancer agents.10–12G. cochlear is medicinally consumed in China. Previous phytochemical investigations, including our recent work, have resulted in the identification of triterpenoids and aromatic meroterpenoids.13–17 In our continuing effort aimed at the isolation of bioactive triterpenoids, we investigated the fruiting bodies of G. cochlear and eighteen triterpenoids (Fig. 1), including ganocochlearic acid A (1), a hexanorlanostane triterpenoid with a γ-lactone ring and a five-membered carbon ring, three 3,4-seco-lanostane triterpenoids (2, 3 and 6), and eight new lanostane triterpenoids (7–12), as well as six known analogues (13–18) were isolated. In addition, compounds 1–13 were evaluated for their cytotoxic activities against five human tumor cell lines by MTS method.
 | ||
| Fig. 1 Chemical structure of compounds 1–18. | ||
Results and discussion
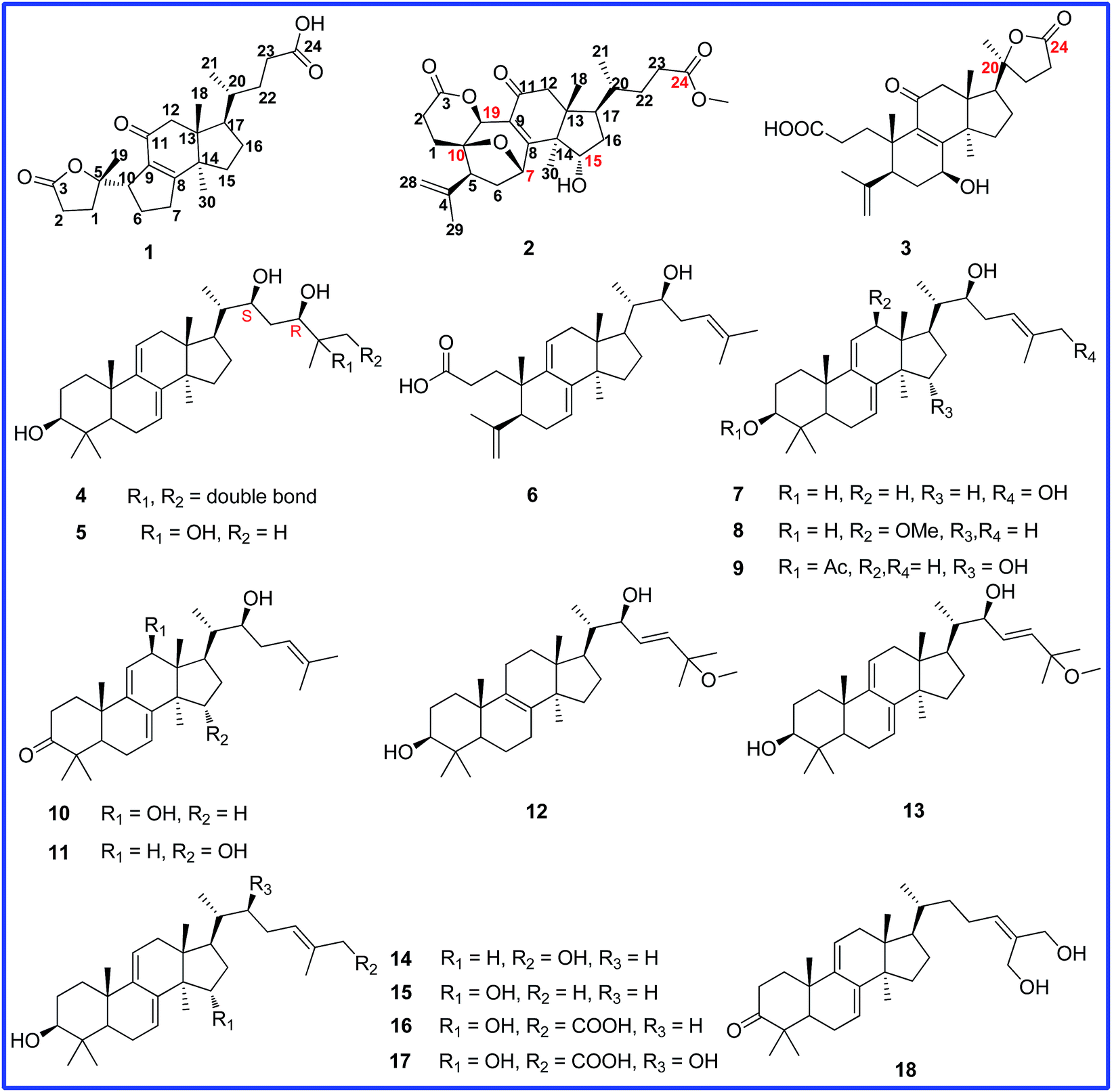
Ganocochlearic acid A (1) was obtained as white powder. Its molecular formula C24H34O5 was deduced from the HRESIMS [M + Na]+ ion at m/z 425.2307 (calcd 425.2304), corresponding to eight degrees of unsaturation. The IR spectrum showed the presence of hydroxyl (3439 cm−1) and α,β-unsaturated carbonyl (1709 cm−1) groups. The 1H NMR spectrum showed three singlet methyl signals at δ 1.48, δ 1.09 and δ 0.78, as well as a doublet methyl signal at δ 0.89 (d, J = 6.6 Hz). The 13C NMR spectrum displayed twenty-four carbon resonances, including four methyls, nine methylenes, three methines and eight quaternary carbons (one ketone carbonyl, one carboxyl, two sp2 quaternary and one oxyquaternary carbons), suggesting that compound 1 was a lanostane-type nortriterpenoid and had the same rings C, D and side chain as fornicatin B,18 which were supported by the 2D NMR spectral analysis (Fig. 2).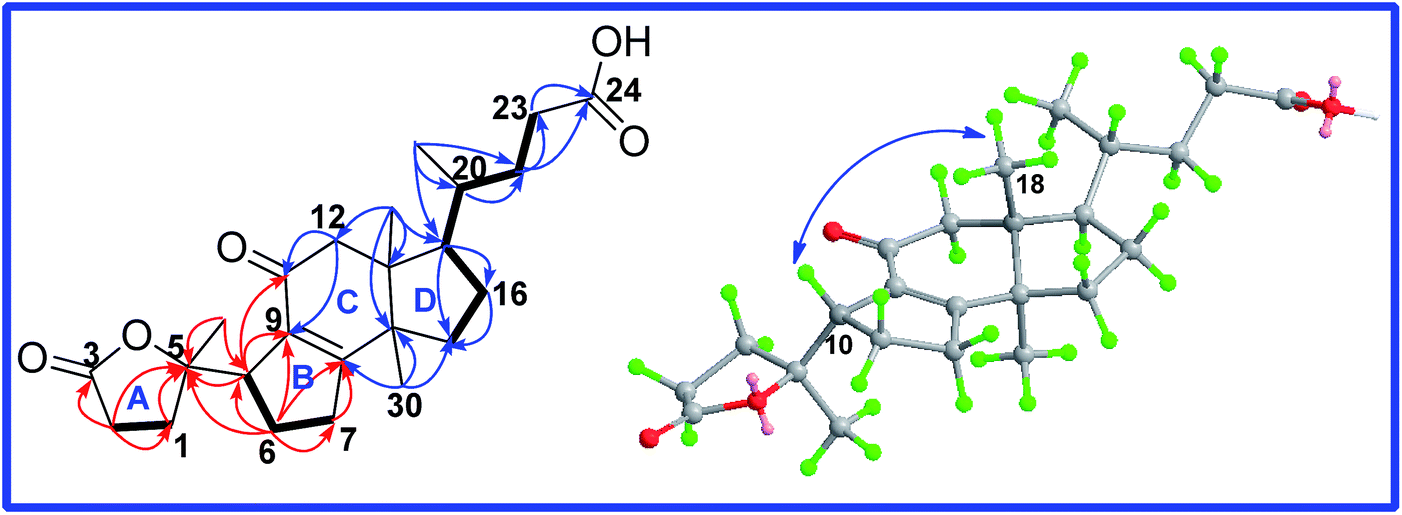 | ||
Fig. 2 Key HMBC ( ), 1H–1H COSY ( ), 1H–1H COSY ( ) and ROESY ( ) and ROESY ( ) correlations of ganocochlearic acid A (1). ) correlations of ganocochlearic acid A (1). | ||
The comparative analysis of its 13C NMR (C5D5N) data with those of fornicatin B showed the strong downfield shift of C-8 (δ 164.1 for fornicatin B; δ 179.3 for 1) and the highfield shift of C-9 (δ 135.4 for fornicatin B; δ 133.0 for 1) and C-11 (δ 199.8 for fornicatin B; δ 198.1 for 1), indicating that ring B of 1 should be different from that of fornicatin B. These differences were proved by the HMBC correlations of H-10 (δ 3.11, m), H2-6 (δ 2.31, m; δ 1.97, m) and H2-7 (δ 2.86, m; δ 2.32, m) to C-8 and C-9; of H-10 to C-11, together with the 1H–1H COSY correlations of H-10/H2-6/H2-7 (Fig. 2). Thus, we unambiguously deduced that ring B of 1 was a five-membered carbon ring.
Apart from seven degrees of unsaturation occupied by three rings, a carboxyl group, a carbonyl group, a double bond and a ester carbonyl group, the remaining one degree of unsaturation was assumed to be representative of another ring (ring A). The HMBC correlations (Fig. 2) of H-10 and H2-6 to C-5 (δ 90.7), of H3-19 (δ 1.50, s), H2-1 (δ 2.70, m; δ 1.86, m) and H2-2 (δ 1.74, m; δ 1.45, m) to C-5 and C-3 (δ 177.4), coupled with the 1H–1H COSY correlation of H2-1/H2-2, deduced ring A of 1 to be a γ-lactone ring. Thus, the planar structure of 1 was established.
The relative orientation of H-10 was determined to be β based on the ROESY correlation of H-10/H3-18. Thus, only two diastereoisomers (1a, 5S,10S; 1b, 5R,10S) remained. The absolute configuration of 1 was established by comparison of the experimental CD spectrum and the calculated ECD data (Fig. 3). The optimized conformations of 1a and 1b were obtained using the time-dependent density functional theory (TD-DFT) method at the B3LYP/6-31+G(d,p) level in the gas phase by using the Gaussian 09 (see ESI†). Further ECD calculations were performed at the PCM-B3LYP/6-31+G(d,p) level in MeOH solution. The result showed that the calculated weighted ECD spectrum of 1a is accordance with the experimental spectrum (Fig. 3). Consequently, the absolute configuration of compound 1 was assigned as 5S,10S.
 | ||
| Fig. 3 Calculated and experimental CD spectra of different diastereomers. (— Calcd.1 for 1a, — Calcd.2 for 1b, — Expl. experimental in MeOH). | ||
Cochlate C (2) was obtained as white powder, and its molecular formula was established as C28H38O7 by HRESIMS ion at m/z 509.2509 [M + Na]+ (calcd 509.2510), indicating 10 degrees of unsaturation. The 1H NMR spectrum showed three singlet methyl signals at δ 1.66, 1.39, 0.89; one doublet methyl signal at δ 0.75 (d, J = 6.0 Hz); one methoxyl signal at δ 3.60 (s); three oxymethine signals at δ 5.40 (d, J = 5.3 Hz), δ 5.21 (s) and δ 4.68 (t, J = 2.4 Hz); as well as one terminal double bond proton signals at δ 4.99 (s), δ 4.87 (s). The 13C-DEPT data (Table 2) displayed the presence of the characteristic signals for the terminal double bond (δ 143.6, C; δ 115.1, CH2), α,β-unsaturated carbonyl group (δ 168.0, C; δ 125.2, C; δ 196.9, C), and two ester carbonyl groups (δ 170.8, C; δ 174.2, C). Its 1D NMR data were similar to those of cochlate A13 except that two oxymethines in 2 replaced two methylenes in cochlate A. The 1H–1H COSY correlations (Fig. 4) between the oxymethine proton (δ 4.68, m) and H-16, between H-16 and H-17, between H-17 and H-20, and between H-20 and H3-21, conjugated with the HMBC correlations of H3-30 to C-13, C-14 and the oxymethine, and of the oxymethine proton to C-13, C-14, C-16, and C-17 indicated that C-15 was bonded to hydroxy group.
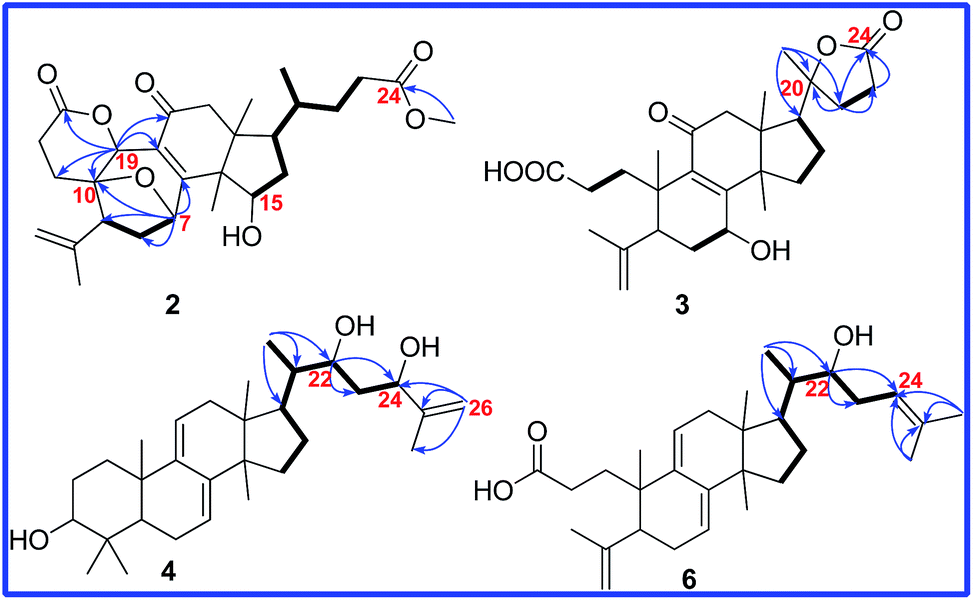 | ||
Fig. 4 Key HMBC ( ) and 1H–1H COSY ( ) and 1H–1H COSY ( ) correlations of compounds 2, 3, 4 and 6. ) correlations of compounds 2, 3, 4 and 6. | ||
Additionally, another oxymethine was assigned to be C-19 on the basis of the HMBC correlations of H-19 (δ 5.21, s) to C-10, C-5, C-8, C-9 and C-11. Moreover, H-19 also showed the HMBC correlation with C-3 (δ 170.8), which deduced that a δ-lactone was between C-3 and C-19. The ROESY correlations (Fig. 5) of H-15/H3-18 and of H-19/H-5/H3-30 proved that the relative orientation of H-15 and H-19 were β and α, respectively. Therefore, the structure of 2 was determined, named cochlate C.
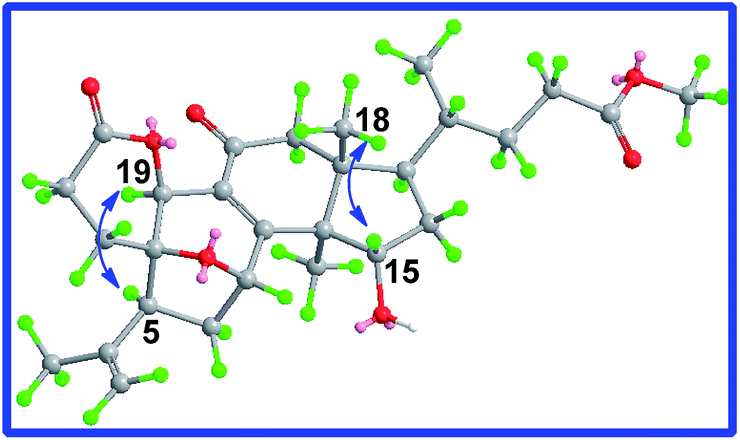 | ||
| Fig. 5 Key ROESY correlations of compound 2. | ||
Compound 3 was isolated as colorless needle crystals. The molecular formula of 3 was established as C27H38O6 by HRESIMS ion at m/z 481.2560 [M + Na]+ (calcd 481.2566). Analysis of 1H 13C, and DEPT spectra (Tables 1 and 2) revealed the presence of a double bond (δ 115.6, CH2; δ 146.4, C), an α,β-unsaturated carbonyl moiety (δ 164.9, C; δ 137.2, C; δ 199.8, C), a carboxyl group (δ 176.4, C), a ester carbonyl group (δ 177.1, C), an oxyquaternary carbon (δ 87.7, C) and an oxymethine (δ 67.7, C), suggesting that compound 3 had the same skeleton as fornicatin B.18 However, comparison of 1D NMR data between 3 and fornicatin B showed five singlet methyls in 3 but four singlet methyls and one doublet methyl in fornicatin B. Meanwhile, a methine in fornicatin B was replaced by the oxyquaternary carbon (δ 87.7) in 3, indicating that C-20 could be linked to an oxygen. Furthermore, the HMBC correlations (Fig. 4) of H3-21 to C-20, C-17, and C-22, of H2-22 to C-20, C-17, C-23, and C-24, and of H2-23 to C-20, C-22, and C-24, along with the analysis of the molecular weight illustrated that a 20 → 24 γ-lactone was present in 3. The planar structure of 3 was finally determined.
| Position | 1a | 2b | 3b | 4a | 5b | 6a | 7b | 8a | 9a | 10a | 11a | 12a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1H (J) | 1H (J) | 1H (J) | 1H (J) | 1H (J) | 1H (J) | 1H (J) | 1H (J) | 1H (J) | 1H (J) | 1H (J) | 1H (J) | |
| a Measured in CDCl3.b Measured in C5D5N. 1H and 13C NMR spectra (δ) were measured at 600 (150) MHz for 1, 4, 6, 8–12; at 500 (125) MHz for 5 and 7; at 400 (100) MHz for 2 and 3. The assignments were based on COSY, HSQC, and HMBC experiments. | ||||||||||||
| 1 | 2.70, m; 1.86, m | 2.71, m | 3.24, m | 1.98, m; 1.43, m | 2.02, m; 1.51, m | 1.83, m; 1.55, m | 2.05, m; 1.52, m | 2.04, m; 1.54, m | 1.97, m; 1.51, m | 2.31, m; 1.78, m | 2.27, m; 1.76, m | 1.72, m; 1.22, m |
| 2 | 1.74, m; 1.45, m | 2.11, m; 1.91, m | 2.67, m | 1.67, m | 1.95, m | 1.26, m; 2.20, m | 1.95, m | 1.78, m; 1.66, m | 1.70, m | 2.34, m; 2.03, m | 2.76, m; 2.35, m | 1.69, m; 1.58, m |
| 3 | 3.25, dd (11.5, 4.0) | 3.46, d (7.7) | 3.46, d (7.8) | 3.27, dd (11.6, 4.2) | 4.50, dd (11.7, 4.3) | 3.24, dd (11.5, 4.0) | ||||||
| 4 | ||||||||||||
| 5 | 2.67, m | 2.50, m | 1.08, m | 1.29, m | 2.23, m | 1.27, m | 1.16, m | 1.54, m | 1.27, m | |||
| 6 | 2.13, m; 1.97, m | 2.31, m | 2.24, m | 2.08, m | 2.18, m | 2.56, m | 2.17, m | 2.09, m | 2.14, m | 2.21, m; 2.07, m | 2.17, m; 2.10, m | 1.67, m; 1.53, m |
| 7 | 2.86, m; 2.32, m | 5.40, d (5.3) | 4.69, t (7.9) | 5.48, d (5.5) | 5.57, d (4.7) | 5.39, br s | 5.57, d (5.0) | 5.58, d (6.5) | 5.85, d (6.5) | 5.56, d (6.6) | 5.90, d (6.1) | 1.72, m; 1.61, m |
| 8 | ||||||||||||
| 9 | ||||||||||||
| 10 | 3.11, d (9.2) | |||||||||||
| 11 | 5.31, d (5.7) | 5.41, d (4.7) | 5.33 br s | 5.41, d (5.0) | 5.67, d (5.5) | 5.31 d (6.0) | 5.23 br s | 5.39, d (5.4) | 2.04, m | |||
| 12 | 2.71, d (16.0); 2.54, d (16.0) | 2.83, d (18.3); 2.60, d (18.3) | 2.89, d (16.3); 2.78, d (16.3) | 2.08, m; 2.25, m | 2.35, d (16.0); 2.20, d (16.0) | 2.19, m; 2.26, m | 2.18, d (18.0); 2.42, d (18.0) | 3.48, br s | 2.35, m; 2.04, m | 4.29, s | 2.35, m; 2.07, m | 1.63, m |
| 13 | ||||||||||||
| 14 | ||||||||||||
| 15 | 2.91, m; 1.73, m | 4.68, m | 3.08, m; 1.70, m | 1.64, m; 1.43, m | 1.75, m; 1.45, m | 1.64, m; 1.33, m | 1.70, m; 1.49 m | 1.65, m; 1.46, m | 4.27, s | 1.75, m; 1.38, m | 4.29, t (6.9) | 1.75, m |
| 16 | 2.10, m; 1.48, m | 2.07, m | 1.82, m; 1.70, m | 1.42, m; 2.10, m | 2.28, m; 1.51, m | 2.04, m | 2.22, m; 1.51, m | 2.06, m; 1.36, m | 2.01, m; 1.77, m | 2.08, m; 1.59, m | 2.01, m; 1.78, m | 2.02, m |
| 17 | 1.80, m | 1.92, m | 2.20, m | 1.39, m | 2.40, m | 1.97, m | 2.45, m | 2.56, m | 2.07, m | 2.15, m | 2.08, m | 1.89, m |
| 18 | 0.81, s | 0.90, s | 1.32, s | 0.57, s | 0.74, s | 0.64, s | 0.73, s | 0.56, s | 0.60, s | 0.57, s | 0.64, s | 0.71, s |
| 19 | 1.50, s | 5.21, s | 1.43, s | 0.97, s | 1.10, s | 0.97, s | 1.09, s | 0.97, s | 1.00, s | 1.23, s | 1.19, s | 0.98, s |
| 20 | 1.48, m | 1.27, m | 1.94, m | 1.68, m | 1.44, dd (6.8, 3.4) | 1.69, m | 1.44, m | 1.38, m | 1.57, m | 1.40, m | 1.49, m | |
| 21 | 0.92, d (6.0) | 0.75, d (6.0) | 1.34, s | 0.91, d (6.0) | 1.21, d (6.0) | 0.89, d (6.8) | 1.24, d (6.5) | 0.93, d (6.8) | 0.87, d (6.3) | 1.09, d (6.3) | 0.88, d (6.6) | 0.87, d (6.8) |
| 22 | 1.35, m; 1.86, m | 1.31 m; 1.82, m | 2.65, m; 2.45, m | 3.99, d (9.9) | 4.43, d (5.7) | 3.68, m | 4.11, m | 3.70, m | 3.59, m | 3.76, m | 3.60, m | 4.30, m |
| 23 | 2.44, m; 2.30, m | 2.38, m; 2.24, m | 2.06, m; 1.79, m | 1.48, m; 1.78, m | 2.05, m; 1.45, m | 2.00, m; 2.29, m | 1.75, m; 1.46, m | 2.29, m | 2.28, m; 1.99, m | 2.03, m | 2.28, m; 1.99, m | 5.61, br s |
| 24 | 4.29, d (9.7) | 4.10, d (9.5) | 5.14, t (7.0) | 6.05, t (6.7) | 5.16, t (7.2) | 5.13, t (7.2) | 5.14, t (7.3) | 5.13, t (6.7) | 5.16 br s | |||
| 25 | ||||||||||||
| 26 | 4.83, s; 5.00, s | 1.53, s | 1.64, s | 4.32, s | 1.64, s | 1.63, m | 1.64, s | 1.64, s | 1.28, s | |||
| 27 | 1.74, s | 1.56, s | 1.72, s | 1.87, s | 1.73, s | 1.73, s | 1.73, s | 1.73, s | 1.28, s (overlap) | |||
| 28 | 4.99, s; 4.87, s | 4.96, s; 5.03, s | 0.88, s | 1.13, s | 4.64, s; 4.67, s | 1.20, s | 1.01, s | 0.88, s | 1.08, s | 1.08, s | 0.80, s | |
| 29 | 1.66, s | 1.80, s | 1.00, s | 1.20, s | 1.66, s | 1.12, s | 0.89, s | 0.94, s | 1.12, s | 1.12, s | 1.00, s | |
| 30 | 1.11, s | 1.40, s | 1.28, s | 0.91, s | 0.96, s | 0.85, s | 0.97, s | 1.07, s | 0.96, s | 0.95, s | 0.96, s | 0.92, s |
| OCH3 | 3.60, s | 3.33, s | 3.16, s | |||||||||
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) CO CO |
2.05, s | |||||||||||
| Position | 1a | 1aa | 2b | 3b | 4a | 5b | 6a | 7b | 8a | 9a | 10a | 11a | 12a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13C | 13C | 13C | 13C | 13C | 13C | 13C | 13C | 13C | 13C | 13C | 13C | 13C | |
| a Measured in CDCl3.b Measured in C5D5N. 1H and 13C NMR spectra (δ) were measured at 600 (150) MHz for 1, 4, 6, 8–12; at 500 (125) MHz for 5 and 7; at 400 (100) MHz for 2 and 3. The assignments were based on COSY, HSQC, and HMBC experiments. | |||||||||||||
| 1 | 32.0 CH2 | 32.0 CH2 | 28.0 CH2 | 32.6 CH2 | 35.4 CH2 | 36.3 CH2 | 34.2 CH2 | 36.3 CH2 | 35.6 CH2 | 35.6 CH2 | 36.5 CH2 | 36.5 CH2 | 35.5 CH2 |
| 2 | 29.9 CH2 | 29.9 CH2 | 26.8 CH2 | 30.8 CH2 | 27.7 CH2 | 28.7 CH2 | 28.3 CH2 | 28.7 CH2 | 27.5 CH2 | 24.5 CH2 | 34.6 CH2 | 34.7 CH2 | 27.8 CH2 |
| 3 | 177.4 C | 177.4 C | 170.8 C | 176.4 C | 78.9 CH | 78.1 CH | 178.3 C | 78.0 CH | 78.7 CH | 80.5 CH | 216.9 C | 216.7 C | 78.9 CH |
| 4 | 143.6 C | 146.4 C | 38.6 C | 39.3 C | 149.1 C | 39.3 C | 38.6 C | 35.3 C | 47.6 C | 47.4 C | 38.8 C | ||
| 5 | 90.7 C | 90.7 C | 48.3 CH | 44.9 CH | 49.0 CH | 49.8 CH | 50.6 CH | 49.7 CH | 48.8 CH | 49.0 CH | 50.6 CH | 50.4 CH | 50.3 CH |
| 6 | 27.0 CH2 | 27.0 CH2 | 38.6 CH2 | 35.3 CH2 | 22.9 CH2 | 23.5 CH2 | 28.3 CH2 | 23.5 CH2 | 22.9 CH2 | 22.7 CH2 | 23.6 CH2 | 23.5 CH2 | 18.2 CH2 |
| 7 | 33.9 CH2 | 33.9 CH2 | 74.2 CH | 67.7 CH | 120.3 CH | 120.9 CH | 117.5 CH | 120.9 CH | 123.3 CH | 121.0 CH | 121.3 CH | 121.0 CH | 27.6 CH |
| 8 | 179.3 C | 179.3 C | 168.0 C | 164.9 C | 142.5 C | 143.0 C | 141.6 C | 143.0 C | 142.6 C | 140.8 C | 141.8 C | 140.9 C | 134.2 C |
| 9 | 133.0 C | 133.0 C | 125.2 C | 137.2 C | 145.8 C | 146.5 C | 136.9 C | 146.5 C | 148.6 C | 145.7 C | 145.5 C | 144.6 C | 134.4 C |
| 10 | 51.8 CH | 51.8 CH | 80.8 C | 41.2 C | 37.3 C | 37.8 C | 38.3 C | 37.8 C | 37.6 C | 37.2 C | 36.9 C | 37.2 C | 36.9 C |
| 11 | 198.1 C | 198.1 C | 196.9 C | 199.8 C | 116.2 CH | 116.6 CH | 120.2 C | 116.6 CH | 113.8 CH | 116.2 CH | 122.0 C | 117.0 CH | 26.4 CH2 |
| 12 | 49.8 CH2 | 49.8 CH2 | 49.6 CH2 | 51.9 CH2 | 37.8 CH2 | 38.3 CH2 | 38.8 CH2 | 38.2 CH2 | 82.6 CH | 38.5 CH2 | 73.9 CH | 38.5 CH2 | 30.9 CH2 |
| 13 | 50.0 C | 49.9 C | 46.6 C | 47.3 C | 43.7 C | 44.0 C | 44.1 C | 44.0 C | 47.3 C | 44.1 C | 49.1 C | 44.1 C | 44.4 C |
| 14 | 50.1 C | 50.1 C | 52.4 C | 51.9 C | 50.2 C | 50.7 C | 50.1 C | 50.7 C | 49.7 C | 51.9 C | 51.5 C | 51.9 C | 39.8 C |
| 15 | 29.7 CH2 | 29.7 CH2 | 70.5 CH | 31.9 CH2 | 31.4 CH2 | 31.9 CH2 | 30.8 CH2 | 31.9 CH2 | 32.5 CH2 | 74.7 CH | 31.5 CH2 | 74.6 CH | 30.8 CH2 |
| 16 | 27.5 CH2 | 27.5 CH2 | 38.6 CH2 | 22.2 CH2 | 27.2 CH2 | 27.7 CH2 | 27.2 CH2 | 27.8 CH2 | 26.8 CH2 | 39.5 CH2 | 27.2 CH2 | 39.4 CH2 | 20.9 CH2 |
| 17 | 49.5 CH | 49.5 CH | 49.0 CH | 53.2 CH | 42.2 CH | 47.6 CH | 47.5 CH | 47.8 CH | 40.1 CH | 45.2 CH | 49.6 CH | 45.2 CH | 46.7 CH |
| 18 | 17.3 CH3 | 17.2 CH3 | 17.7 CH3 | 19.2 CH3 | 15.3 CH3 | 16.0 CH3 | 16.3 CH3 | 16.0 CH3 | 16.7 CH3 | 15.8 CH3 | 10.3 CH3 | 15.9 CH3 | 15.7 CH3 |
| 19 | 27.8 CH3 | 27.8 CH3 | 74.5 CH | 22.2 CH3 | 22.6 CH3 | 23.1 CH3 | 21.9 CH3 | 23.0 CH3 | 22.5 CH3 | 22.8 CH3 | 21.8 CH3 | 22.1 CH3 | 19.4 CH3 |
| 20 | 35.7 CH | 35.7 CH | 35.9 CH | 87.7 C | 46.9 CH | 42.4 CH | 40.2 CH | 41.7 CH | 40.4 CH | 40.1 CH | 38.8 CH | 40.1 CH | 41.9 CH |
| 21 | 18.4 CH3 | 18.2 CH3 | 17.7 CH3 | 25.9 CH3 | 12.1 CH3 | 12.7 CH3 | 11.6 CH3 | 12.4 CH3 | 10.6 CH3 | 11.7 CH3 | 11.9 CH3 | 11.7 CH3 | 11.9 CH3 |
| 22 | 31.0 CH2 | 31.1 CH2 | 31.1 CH2 | 28.0 CH2 | 74.4 CH | 73.6 CH | 73.2 CH | 72.8 CH | 73.6 CH | 73.2 CH | 74.5 CH | 73.2 CH | 73.7 CH |
| 23 | 31.2 CH2 | 31.3 CH2 | 31.1 CH2 | 31.9 CH2 | 40.2 CH2 | 36.8 CH2 | 34.2 CH2 | 34.9 CH2 | 34.1 CH2 | 34.2 CH2 | 35.0 CH2 | 34.3 CH2 | 132.6 CH |
| 24 | 179.0 C | 174.6 C | 174.2 C | 177.1 C | 76.8 CH | 79.6 CH | 120.8 CH | 123.2 CH | 120.9 CH | 120.7 CH | 120.7 CH | 120.6 CH | 134.7 CH |
| 25 | 147.6 C | 72.4 CH | 134.5 C | 137.4 C | 134.0 C | 134.8 C | 134.9 C | 134.9 C | 74.7 C | ||||
| 26 | 110.5 CH2 | 25.8 CH3 | 18.2 CH3 | 68.0 CH2 | 17.9 CH3 | 18.0 CH3 | 18.1 CH3 | 18.1 CH3 | 25.8 CH3 | ||||
| 27 | 17.9 CH3 | 25.9 CH3 | 26.0 CH3 | 14.3 CH3 | 25.7 CH3 | 25.9 CH3 | 26.0 CH3 | 25.8 CH3 | 26.0 CH3 | ||||
| 28 | 115.1 CH2 | 115.6 CH2 | 15.7 CH3 | 16.6 CH3 | 111.8 CH2 | 16.6 CH3 | 28.1 CH3 | 27.9 CH3 | 25.3 CH3 | 25.3 CH3 | 15.4 CH3 | ||
| 29 | 22.4 CH3 | 22.2 CH3 | 28.0 CH3 | 28.8 CH3 | 21.9 CH3 | 28.8 CH3 | 15.7 CH3 | 16.9 CH3 | 22.6 CH3 | 22.4 CH3 | 27.9 CH3 | ||
| 30 | 23.8 CH3 | 23.9 CH3 | 19.1 CH3 | 27.8 CH3 | 25.5 CH3 | 26.3 CH3 | 23.9 CH3 | 25.9 CH3 | 28.5 CH3 | 17.1 CH3 | 24.9 CH3 | 17.0 CH3 | 24.3 CH3 |
| OCH3 | 51.8 CH3 | 51.3 CH3 | 55.2 CH3 | 50.4 CH3 | |||||||||
| CO |
21.3 CH3 | ||||||||||||
| 170.9 C | |||||||||||||
The ROESY correlation of H-7/H3-30 deduced 7-OH to be β. The absolute configuration of C-20 was determined to be S on the basis of the X-ray crystal diffraction analysis (Fig. 6). Accordingly, the structure of 3 was established to be (20S)-7β-hydroxy-11-oxo-3,4-seco-25,26,27-trinorlanosta-4(28),8-dien-3-oic-20 → 24 lactone and named as cochlearic acid A (3).
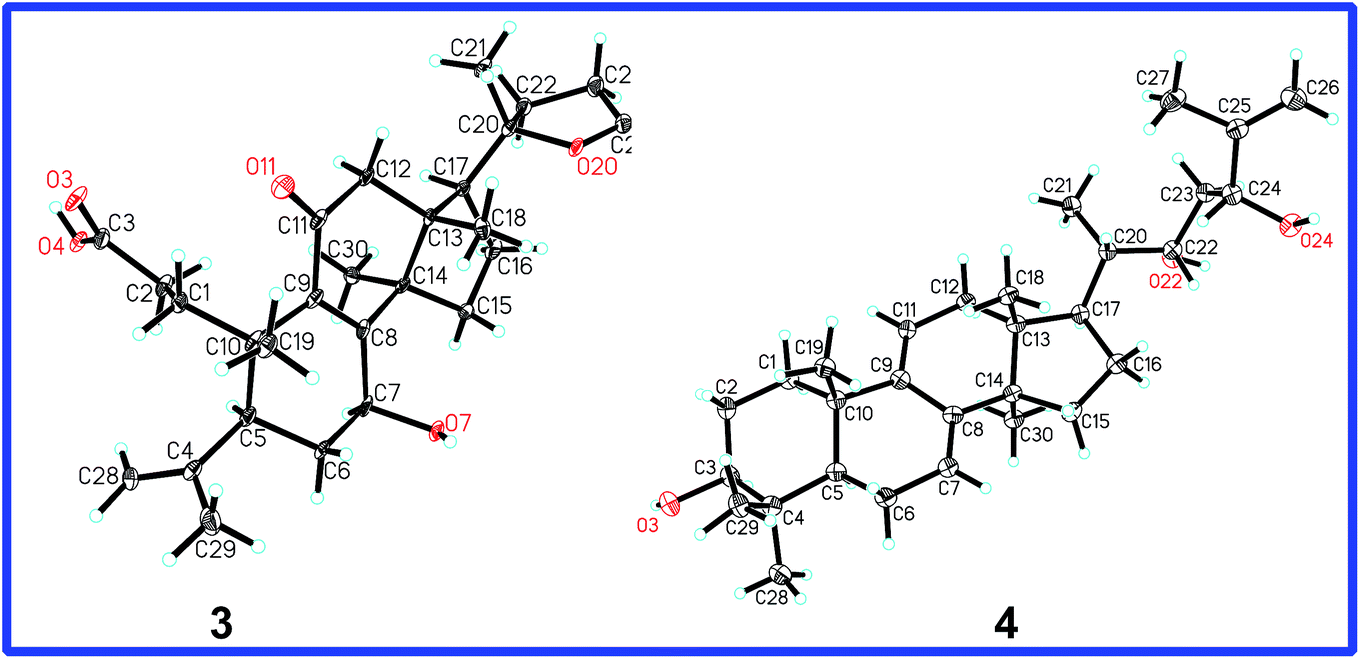 | ||
| Fig. 6 X-ray structures of compounds 3 and 4. | ||
Compound 4 was isolated as colorless needle crystals with a molecular formula of C30H48O3 based on HREIMS ion at m/z 456.3612 [M]+ (calcd 456.3603). The 1D NMR spectroscopic data (Tables 1 and 2) showed the following signals: six singlet methyls (δ 1.74, δ 1.00, δ 0.97, δ 0.91, δ 0.87 and δ 0.56); one doublet methyl (δ 0.91, d, J = 6.0 Hz); eight methylenes, including one sp2 methylene (δ 4.83, s; δ 5.00, s); eight methines, including three oxymethines (δ 3.25, dd, J = 11.5 and 4.0 Hz; δ 3.99, d, J = 9.9 Hz; δ 4.29, d, J = 9.7 Hz) and two sp2 methines [δ 5.48 (d, J = 5.5 Hz), δ 5.31 (d, J = 5.7 Hz)]; as well as seven quaternary carbons (three sp2 quaternary carbons). These data indicated that the structural skeleton of compound 4 was a lanosta-7,9(11)-dien-3-ol, which was supported by the 2D NMR spectra (Fig. 4).
The 1H–1H COSY correlations between H3-21 and H-20, between H-22 (δ 3.99, d, J = 9.9 Hz) and H2-23, between H2-23 and H-24 (δ 4.29, d, J = 9.7 Hz), together with the HMBC correlations of H3-21 to C-20, C-17 and C-22 (δ 74.4), of H-22 to C-20, C-24 (δ 76.8) and C-25 (δ 147.6), of H-23 to C-22, C-24, C-25, and C-26 (δ 110.5), of H3-27 to C-24, C-25 and C-26, indicated that compound 4 had the same side chain moiety (22,24-dihydroy-25-ene) as inontsutriol D.19 The stereocenters (C-22 and C-24) of the side chain moiety in 4 were confirmed by the X-ray crystal diffraction analysis. A single X-ray crystallographic analysis (Cu Kα radiation, Fig. 6) of 4 assigned C-22 and C-24 as S and R, respectively. Thus, the structure of 4 was determined to be lanosta-7,9(11),25-trien-3β,22S,24R-triol and named as ganodercochlearin D (4).
Compound 5 was found to possess the molecular formula C30H50NaO3, as evidenced by its positive HRESIMS. Its 1D NMR data (Tables 1 and 2) closely resembled those of 4. The main difference observed in the 13C NMR spectra was that the signals corresponding to the terminal double bond (δ 147.6, δ 110.5) at C-25 and C-26 were replaced by an oxymethine (δ 72.4, CH) and a methyl (δ 25.8 CH3), respectively. This difference was confirmed by the HMBC correlations of H-24 (δ 4.10, d, J = 9.5 Hz) with C-25 (δ 72.4), C-26 (δ 25.8) and C-27 (δ 25.9), of H3-26 (δ 1.53, s) and H3-27 with C-24 (δ 79.6) and C-25. Therefore, the structure of 5 was established to be lanosta-7,9(11)-3β,22S,24R,25-tetraol, named ganodercochlearin E (5).
Based on the positive HREIMS of 6, its molecular formula C30H46O3 was deduced, containing eight degrees of unsaturation. Its 13C NMR spectrum showed thirty carbon resonances. Among them, the signals at δ 117.5, δ 141.6, δ 136.9 and δ 120.2 were characterized for the conjugated double bond (Δ7 and Δ9,11). Meanwhile, the signals at δ 149.1, C; δ 111.8, CH2 and δ 178.3, C were assigned as the terminal double bond (C-4, C-28) and the carboxyl group (C-3) on the basis of the DEPT, HSQC and HMBC spectra. These informations indicated that compound 6 had the same cyclic structure as 16-deoxyporicoic acid B20 with a 3,4-seco-7,9(11)-dien-lanostane skeleton. Additionally, one double bond signals (δ 120.8, CH; δ 134.5, C) and one oxymethine signal (δ 73.2, CH) were also observed in the 13C NMR spectrum of 6. Moreover, the HMBC correlations of H3-21 (δ 0.89, d, J = 6.8 Hz) with C-17 (δ 47.5), C-20 (δ 40.2), and C-22 (δ 73.2), of H-22 (δ 3.68, m) with C-17, C-20, C-21, C-23, and C-24, together with the 1H–1H COSY correlations of H3-21/H-20/H-22/H-23/H-24, confirmed that the side chain of compound 6 resembled that of compound 5, except for the replacement of one oxymethine (C-24) and one oxyquaternary carbon (C-25) in 5 by two sp2 olefinic carbons in 6. Accordingly, the structure of 6 was assigned as (22S)-hydroxy-3,4-seco-lanosta-4(28),7,9(11),24-tetraen-3-oic acid and named as cochlearic acid B (6).
The molecular formula of ganodercochlearin F (7) was deduced from the HRESIMS ion at m/z 479.3493 [M + Na]+ (calcd 479.3501). Its 1D NMR spectroscopic data (Tables 1 and 2) and the DEPT analysis of 7 showed that this compound bore a close resemblance to compound 5. However, the presence of the signals at δ 72.8 (CH), δ 123.2 (CH), and δ 137.4 (C), conjugated with a set of 2D NMR correlations indicated that compound 7 had the same side chain moiety as compound 6. Further comparison of the 13C-DEPT data between 6 and 7 displayed that an oxymethylene in 7 replaced a methyl in 6. The HMBC correlations (Fig. 4) of the oxymethylene protons (δ 4.32, s) with C-24, C-25 and C-27 were observed, which indicated that the hydroxyl group was linked to C-26.
The geometry of Δ24 were determined to be Z on the basis of the ROESY correlation of H-24/H2-26. Thus, the structure of 7 was established to be (22S)-lanosta-7,9(11),24Z-triene-3β,22,26-triol and named as ganodercochlearin F (7).
Ganodercochlearin G (8) possessed a molecular formula of C31H50O3 as determined by HREIMS (m/z 470.3755 [M]+, calcd 470.3760). The signals in the 1H and 13C NMR spectroscopic data (Tables 1 and 2) of 8 strikingly matched those of 7, indicating that they shared the same backbone. The major difference were the absence of the oxymethylene and the presence of an oxymethine and a methoxyl group in 8. The key HMBC correlations of H3-18 with the C-13, C-14, C-17, and the oxymethine (δ 82.6); of the oxymethine proton (δ 3.48, br s) with C-9, C-11, C-13 and C-14, as well as of the methoxyl protons (δ 3.33, s) with C-12, which indicated that the methoxyl group was linked to C-12.
12β-OMe was deduced by the ROESY correlation of H-12/H3-30. Therefore, the structure of 8 was assigned as (22S)-12β-methoxyl-lanosta-7,9(11),24-triene-3β,22-diol, named ganodercochlearin G (8).
The molecular formula of ganodercochlearin H (9) was determined to be C32H50O4 from the HREIMS ion at m/z 498.3715 [M]+, calcd 498.3709. The 1D NMR data (Tables 1 and 2) of 9 obviously showed the presence of an acetyl (δ 2.05, s; δ 21.3, CH3; δ 170.9, C). On the basis of the HMBC correlation of H-3 with the acetyl carbonyl carbon (δ 170.9), this acetoxyl group was located at C-3. In addition, by comparison of the 1D NMR spectra of 9 and 7, an oxymethine (δ 74.7) was observed in 9 instead of the oxymethylene in 7. The obvious HMBC correlations of H3-30 with C-13, C-14, C-8, and the oxymethine, of the oxymethine proton (δ 4.27, s) with C-13, C-14, C-30, C-16, and C-17 indicated that the oxymethine was located at C-15. Moreover, the ROESY correlation of H-15/H3-18 assigned 15-OH as α-orientation. Accordingly, the structure of 9 was established to be (22S)-3β-acetoxyl-lanosta-7,9(11),24-trien-15α,22-diol, named ganodercochlearin H (9).
Ganodercochlearin I (10) and ganodercochlearin J (11) gave the same molecular formula, C30H46O3 (HREIMS). Both of them had thirty carbon resonances, including eight methyls, six methylenes, eight methines (three sp2 methines and two oxymethines), and eight quaternary carbons (one carbonyl and three sp2 quaternary carbons). These signals showed that their backbone was 22-hydroxy-lanosta-7,9(11),24-triene-3-one. This was supported by the 2D NMR analysis. Thus, the only significant difference between 10 and 11 could be the position of another oxymethine (δ 73.9 for 10; δ 74.6 for 11). The HMBC correlations of the oxymethine proton (δ 4.29, s) in 10 with C-11, C-13, and C-18, as well as of the oxymethine (δ 4.29, t, J = 6.9 Hz) in 11 with C-13, C-14, C-30, C-16, and C-17 confirmed that C-12 in 10 and C-15 in 11 were connected to the hydroxyl group, respectively. Similarly, on the basis of the ROESY correlations of H-12/H3-30 and of H-15/H3-18, 12-OH and 15-OH were assigned as β-orientation and α-orientation, respectively. Therefore, the structures of 10 and 11 were determined to be (22S)-12β,22-dihydroxy-lanosta-7,9(11),24-triene-3-one and (22S)-15α,22-dihydroxy-lanosta-7,9(11),24-triene-3-one, named ganodercochlearins I and J (10 and 11).
Ganodercochlearin K (12) possessed a molecular formula of C31H52O3 as determined by HREIMS ion at m/z 472.3924 [M]+ (calcd 472.3916). The 1D NMR data (Tables 1 and 2) of 12 were similar to those of ganodercochlearin C (13), except for the absence of the conjugated double bonds (Δ7 and Δ9,11) and the presence of a double bond between C-8 and C-9, which was confirmed by the HMBC correlations of H2-6 and H2-7 with C-8 (δ 134.2), of H2-11 with C-8 (δ 134.2, C), C-9 (δ 134.4, C) and C-10. The structure of 12 was finally assigned as (22S)-25-methoxyl-lanosta-8,23-dien-3β,22-diol and named as ganodercochlearin K (12).
The six known lanostane triterpenoids were identified by comparison of experimental and literature spectroscopic data as ganodercochlearin C (13),13 ganodermadiol (14),21 polycarpol (15),22 ganoderic acid C2 (16),23 3β,15α,22β-trihydroxylanosta-7,9(11),24-trien-26-oic acid (17),24 and ganoderiol F (18).25
Ganocochlearic acid A (1) is a hexnorlanostane triterpenoid possessing a γ-lactone ring (A ring) and a five-membered carbon ring (B ring). A ring is connected with B ring through a single bond (C-5–C-10). Detailed comparison of the structures between fornicatin B and 1, it is found that they have the same C, D rings and the side chain moiety. Thus, we deduce that compound 1 could derive from fornicatin B via the degradation of C-4, C-28 and C-29, Baeyer–Villiger reaction and Wagner–Meerwein rearrangement. A possible biosynthetic pathway is proposed and shown in Scheme 1.
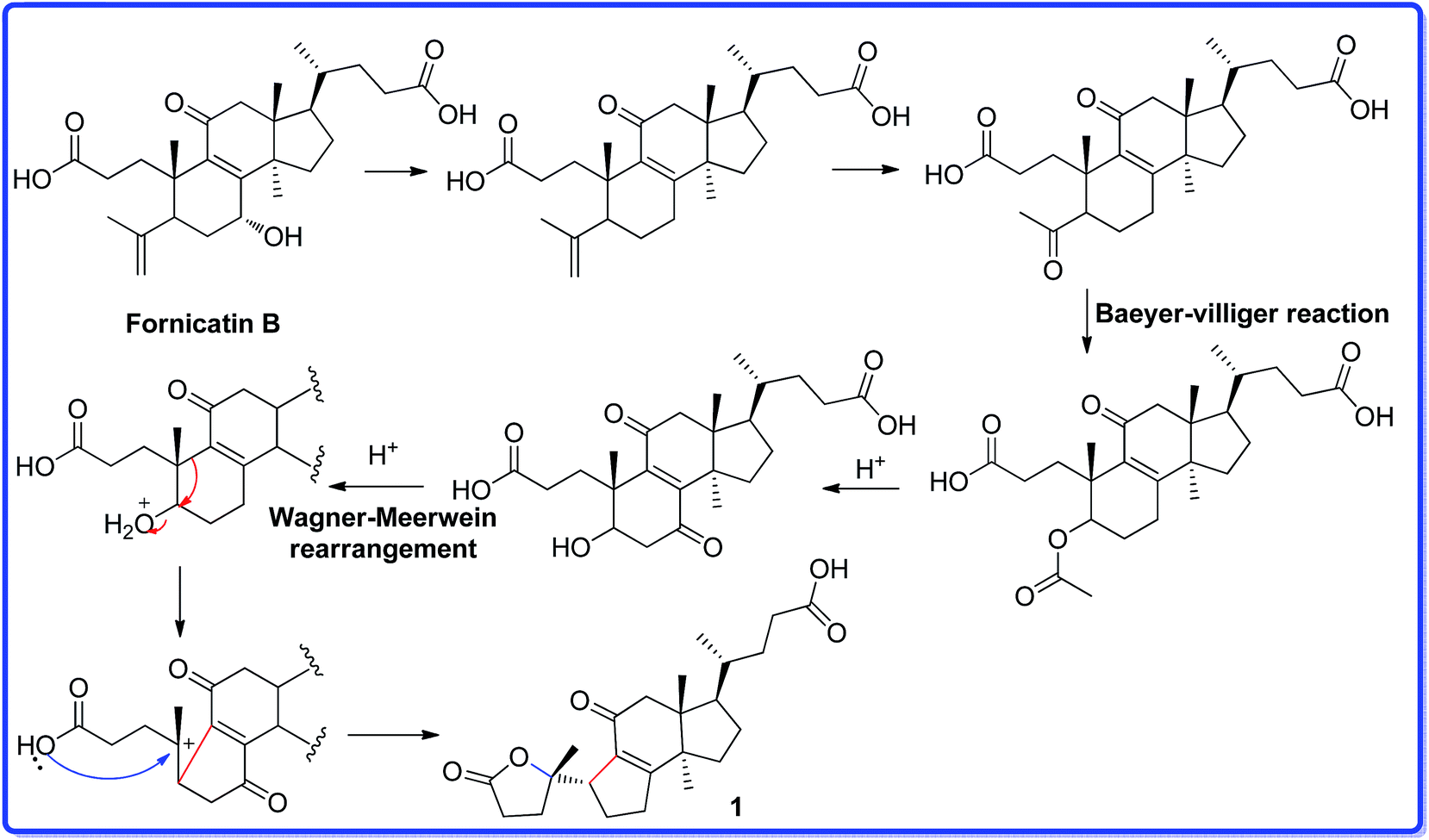 | ||
| Scheme 1 The plausible biosynthetic pathway for 1. | ||
Cytotoxic effects of compounds 1–13 were tested against five human tumor cell lines (HL-60, SMMC-7721, A-549, MCF-7, and SW480) by MTS method. Compounds 7–9, 11–13 had moderate cytotoxic activities against five cell lines with IC50 values ranging from 8 to 30 μM (Table 3). Compound 4 exhibited cytotoxic activity against MCF-7 and SW480 cancer cells with respective IC50 value of 9.15 and 15.9 μM, while the positive control, cisplatin (DDP), showed the activity toward these cancer cells with IC50 values of 12.7 and 12.5 μM. Compound 10 displayed weak inhibition against HL-60, SMMC-7721, A-549 and MCF-7 cells (IC50: 32.9, 28.1, 38.5 and 25.1 μM). In the other hand, 3,4-seco-lanostane triterpenoids (1–3, 6) exhibited no inhibitory effects against all the test human tumor cells. Ganodercochlearin D (4) and ganodercochlearin E (5) had the same structure, nevertheless, compound 5 show weak cytotoxicity with IC50 values of >40 μM, suggesting that the terminal double bond in the side chain of 4 could be the key structural feature. The results are shown in Table 3.
| Compounds | HL-60 | SMMC-7721 | A-549 | MCF-7 | SW480 |
|---|---|---|---|---|---|
| a DDP = cis-dichlorodiamineplatinum(II) or cisplatin, a positive control. | |||||
| 4 | 6.91 ± 1.02 | 10.5 ± 2.11 | 12.1 ± 2.72 | 9.15 ± 1.23 | 15.9 ± 3.11 |
| 7 | 16.3 ± 3.34 | 17.2 ± 3.63 | 16.5 ± 3.15 | 16.8 ± 3.43 | 17.8 ± 4.01 |
| 8 | 13.9 ± 2.84 | 17.0 ± 3.36 | 15.8 ± 3.24 | 17.4 ± 3.77 | 17.1 ± 3.72 |
| 9 | 16.2 ± 3.32 | 16.8 ± 3.21 | 14.7 ± 2.83 | 15.8 ± 3.45 | 15.9 ± 3.45 |
| 10 | 32.9 ± 4.32 | 28.1 ± 4.25 | 38.5 ± 4.83 | 25.1 ± 4.26 | >40 |
| 11 | 14.7 ± 2.32 | 17.6 ± 3.22 | 26.8 ± 4.53 | 19.3 ± 3.91 | 22.3 ± 4.10 |
| 12 | 13.6 ± 2.71 | 16.3 ± 3.02 | 16.9 ± 3.03 | 21.6 ± 3.81 | 19.2 ± 3.72 |
| 13 | 19.3 ± 3.81 | 10.1 ± 2.12 | 8.84 ± 1.47 | 13.4 ± 2.22 | 15.9 ± 2.83 |
| DDP | 1.14 ± 0.23 | 4.63 ± 0.55 | 4.68 ± 0.56 | 12.7 ± 2.31 | 12.5 ± 2.34 |
3,4-seco-Norlanostane and 7,9(11),24-trien-lanostane triterpenoids were widely distributed in the fruiting bodies of G. cochlear. Indeed, these kinds of triterpenoids were also found in other Ganoderma species and exhibited various bioactivities. 3,4-seco-Nortriterpenoids, fornicatins A, D, and F, lowered the ALT and AST levels in HepG2 cells treated with H2O2.13 Meanwhile, 7,9(11),24-trien-lanostane triterpenoids showed antitumor, antiviral, antifungal (Fitoterapia, 2003, 74, 375), and antimycobacterial activities.26–30 Previous and present researches indicated that lanostane triterpenoids play an important role in explaining the wide application of Ganoderma, and deserve further research.
Conclusions
In conclusion, twelve new lanostane triterpenoids, including a hexanorlanostane triterpenoid (1), three 3,4-seco-lanostane triterpenoids (2, 3, 6), and nine 7,9(11),24-trien-lanostane triterpenoids (4, 5, and 7–12), along with six known analogues (13–18) were isolated from the fruiting bodies of G. cochlear. Among of them, compounds 4, 7–13 showed cytotoxic activities.Experimental section
General experimental procedures
Optical rotations were obtained with a Jasco P-1020 polarimeter. UV spectra was recorded using Shimadzu UV2401PC spectrophotometer. 1H and 13C NMR spectra were measured on Bruker AV-400 and DRX-500 instruments (Bruker, Zurich, Switzerland) using TMS as internal standard for chemical shifts. Chemical shifts (δ) are expressed in ppm with reference to the TMS resonance. ESIMS and HRTOF-ESIMS data were recorded on an API QSTAR Pulsar spectrometer. EIMS and HRTOF-EIMS data were taken on an Waters Auto Spec Premierp 776 spectrometer (America, Waters). Infrared spectra were recorded on a Bruker Tensor-27 instrument by using KBr pellets. An Agilent 1100 series instrument equipped with an Agilent ZORBAX SB-C18 column (5 μm, 9.4 mm × 250 mm) was used for high-performance liquid chromatography (HPLC) analysis.Fungal material
The fruiting bodies of G. cochlear were purchased in July 2013 from Traditional Chinese Medicine Market in Kunming. The mushroom was identified by Prof. Liu Peigui, Kunming Institute of Botany, Chinese Academy of Science. A specimen (no. 13071501) was deposited in the Herbarium of the Department of Taxonomy, Kunming Institute of Botany, Chinese Academy of Sciences.Extraction and isolation
G. cochlear (68 kg) mushrooms were chipped and extracted with 95% EtOH under reflux three times (three hours per time). The combined ethanol extracts were evaporated under reduced pressure. The residue was suspended in H2O and extracted with EtOAc. The volume of the combined EtOAc extracts was reduced to one-third under reduced pressure. The residue was fractionated by macroporous resin (D-101; MeOH–H2O, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, 70:30 and 90:10, v/v): fractions I–III.
:50, 70:30 and 90:10, v/v): fractions I–III.
We taken out 100 g of fraction II and used the reversed-phase (RP-18) silica gel chromatography to treat it and obtain eight subfractions (MeOH–H2O: 50:50 → 80:20, Fr. II-1 → Fr. II-8). Every fractions was treated by Sephadex G-25 (MeOH) to three parts. Fr. II-4-2 was purified by preparative TLC (P-TLC, CHCl3–MeOH–acetic acid: 80:1:1) to give compounds 1 (10 mg) and 2 (12 mg). Compound 3 (22 mg) was obtained from Fr. II-4-3 by recrystallization (MeOH). Fr. II-6-2 was absorbed to column chromatography (CC, silica gel) and eluted with CHCl3–acetone: 80:1, 60:1, 40:1 to give four subfractions (Fr. II-6-2a → Fr. II-6-2d). Fr. II-6-2b was purified by P-TLC (CHCl3–MeOH: 50:1) to gain ganodermadiol (14, 13 mg) and polycarpol (15, 19 mg). Four subfractions (Fr. II-7-2a → Fr. II-7-2d) were obtained from Fr. II-7-2 by CC (silica gel, CHCl3–acetone, step gradient). Fr. II-7-2a was treated by the recrystallization to obtain compound ganodermatriol (18, 21 mg). Similarly, compound 5 (12 mg) was isolated from the Fr. II-7-2b by CC (silica gel). Compound 7 (7 mg) was obtained from the Fr. II-7-2c by P-TLC. Fr. II-8-2 was treated by the P-TLC to gain ganoderic acid C2 (16, 12 mg) and 3β,15α,22β-trihydroxylanosta-7,9(11),24-trien-26-oic acid (17, 23 mg).
Fraction III (1.5 kg) was treated by column chromatography (CC, silica gel; CHCl3–MeOH step gradients) to give four subfractions (80:1, 50:1, 20:1 and 5:1, III-1–III-4). Fraction III-2 (50 g) was further fractionated by RP-18 to obtain ten subfractions (MeOH–H2O: 50:50 → 80:20). On the basis of the TLC results, Fr. III-2-3 and Fr. III-2-6 were chosen for the further separation.
Fr. III-2-3 was separated by the RP-18 (MeOH–H2O, 50:50 → 80:20, v/v) to obtain seven subfractions (Fr. III-2-3a → Fr. III-2-3g). Compound 6 (23 mg) was isolated from the Fr. III-2-3b, which was treated by the Sephadex G-25 (MeOH) and recrystallization (MeOH). Fr. III-2-3c was absorbed to Sephadex G-25 and eluted with MeOH to give four fractions. Fr. III-2-3c-2 was further treated by the preparative TLC (P-TLC, CHCl3–MeOH: 50:1) to give compounds 4 (25 mg) and 10 (18 mg). Compound 8 (35 mg) was obtained from Fr. III-2-3 by CC (silica gel; CHCl3–MeOH) and P-TLC. Fr. III-2-3f was further separated by reversed-phase HPLC (CH3CN–H2O, 85%) to afford compound 12 (24 mg) and ganodercochlearin C (13, 30 mg). By repeated CC, HPLC separation, and recrystallization (MeOH/H2O), from fraction III-2-3g, compounds 9 (12 mg) and 11 (21 mg) were isolated. The spectroscopic data of the new compounds (1–12) are listed herein.
ε) nm: 258 (3.69), 202 (3.72) nm; IR (KBr) νmax: 3439, 2969, 2934, 2881, 1709, 1671 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; ESIMS m/z 425 [M + Na]+, HREIMS m/z 402.2401 [M]+ (calcd for C24H34O5, 402.2406).ε) nm: 257 (3.92) nm; IR (KBr) νmax: 3430, 2975, 1711, 1676 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; ESIMS m/z 509 [M + Na]+, HRESIMS m/z 509.2509 [M + Na]+ (calcd for C28H38NaO7, 509.2515).ε) nm: 257 (3.87), 227 (3.66) nm; IR (KBr) νmax: 3443, 2987, 1706, 1656 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; ESIMS m/z 457 [M − H]−, HRESIMS m/z 481.2560 [M + Na]+ (calcd for C27H38NaO6, 481.2561).ε) nm: 242 (4.18), 199 (3.80) nm; IR (KBr) νmax: 3443, 2987, 1706, 1656 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; EIMS m/z 456 [M]+, HREIMS m/z 456.3612 [M]+ (calcd for C30H48O3, 456.3603).475 reflections measured, 4453 independent reflections (Rint = 0.0469). The final R1 values were 0.0597 (I > 2σ(I)). The final wR(F2) values were 0.1645 (I > 2σ(I)). The final R1 values were 0.0598 (all data). The final wR(F2) values were 0.1646 (all data). The goodness of fit on F2 was 1.089. Flack parameter = 0.0(3). The Hooft parameter is −0.04(7) for 2035 Bijvoet pairs.ε) nm: 245 (4.12) nm; IR (KBr) νmax: 3385, 2973, 2961, 2843, 1465 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; ESIMS m/z 497 [M + Na]+, HRESIMS m/z 497.3603 [M + Na]+ (calcd for C30H50NaO4, 497.3607).ε) nm: 240 (4.06) and 202 (4.12) nm; IR (KBr) νmax: 3385, 2973, 2961, 2843, 1465 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; EIMS m/z 454 [M]+, HREIMS m/z 454.3441 [M]+ (calcd for C30H46O3, 454.3447).ε) nm: 245 (3.98) nm; IR (KBr) νmax: 3385, 2973, 2961, 2843, 1465 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; ESIMS m/z 497 [M + Na]+, HRESIMS m/z 479.3493 [M + Na]+ (calcd for C30H48NaO3, 479.3501).ε) nm: 246 (3.03) and 201 (3.89) nm; IR (KBr) νmax: 3385, 2973, 2961, 2843, 1465 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; EIMS m/z 470 [M]+, HREIMS m/z 470.3755 [M]+ (calcd for C31H50O3, 470.3760).ε) nm: 242 (4.05) and 201 (3.88) nm; IR (KBr) νmax: 3385, 2973, 2961, 2843, 1465 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; EIMS m/z 498 [M]+, HREIMS m/z 498.3715 [M]+ (calcd for C32H50O4, 498.3709).ε) nm: 245 (4.11) and 201 (3.94) nm; IR (KBr) νmax: 3385, 2973, 2961, 2843, 1465 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; EIMS m/z 454 [M]+, HREIMS m/z 454.3440 [M]+ (calcd for C30H46O3, 454.3447).ε) nm: 242 (4.24) and 201 (4.17) nm; IR (KBr) νmax: 3385, 2973, 2961, 2843, 1465 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; EIMS m/z 454 [M]+, HREIMS m/z 454.3441 [M]+ (calcd for C30H46O3, 454.3447).ε) nm: 222 (3.41) and 200 (4.12) nm; IR (KBr) νmax: 3385, 2973, 2961, 2843, 1465 cm−1; 1H and 13C-DEPT data: see Tables 1 and 2; EIMS m/z 472 [M]+, HREIMS m/z 472.3924 [M]+ (calcd for C31H52O3, 472.3916).X-ray diffraction analysis of 3 and 4
The crystal structure of 3 and 4 were solved by direct method SHELXS-97 (Sheldrich, G. M. University of Gottingen; Gottingen, Germany, 1997) and the full-matrix least-squares deposited in the Cambridge Crystallographic Data Centre (deposition number: 963836 for 3; 1405439 for 4).†Computational methods
Quantum chemical method was used to assign the absolute configuration of compound 1 by comparing the experimental and calculated electronic circular dichroism (ECD) spectra at time-dependent density functional theory (TDDFT). Conformational analysis was initially carried out by using Discovery Studio 4.1 Client conformational searching and molecular mechanics methods (MMFF94). The selected conformers were then optimized at the B3LYP/6-31+G(d,p) level in the gas phase by using the Gaussian09.31 Further ECD calculations were performed at the PCM-B3LYP/6-31+G(d,p) level in MeOH solution.Cytotoxicity assay
The cytotoxicity of the tested compounds was evaluated using the MTS assay.32 Briefly, 1 × 105 cells per mL from five human cancer cell lines, breast cancer MCF-7, hepatocellular carcinoma SMMC-7721, human myeloid leukemia HL-60, lung cancer A-549 cells, and colon cancer SW480 were seeded in 96-well plates. After 24 h of incubation, the cells were treated with or without test compounds or cisplatin (DDP, positive control) at a given concentrations for 48 h. MTS was then added to each well, and the plates were stored for 4 h. Absorbance was read at 490 nm. Cytotoxicity is reported as percent of control, where 100% is the viability of cells without test compounds.Acknowledgements
The project was financially supported by the General Program of NSFC (No. 81172940) and Knowledge Innovation Program of the CAS (Grant No. KSCX2-YW-G-038, KSCX2-YW-R-194), Top Talents Program from the Department of Science and Technology in Yunnan Province (20080A007), as well as Foundation of State Key Laboratory of Phytochemistry and Plant Resources in West China (P2010-ZZ14).Notes and references
- J. L. Rios, I. Andujar, M. C. Recio and R. M. Giner, J. Nat. Prod., 2012, 75, 2016–2044 CrossRef CAS PubMed.
- H. B. Hu, N. S. Ahn, X. L. Yang, Y. S. Lee and K. S. Kang, Int. J. Cancer, 2002, 102, 250–253 CrossRef CAS PubMed.
- J. P. Zhang, L. M. Zheng, J. H. Wang, K. E. Magnusson and X. Liu, Phytother. Res., 2009, 23, 844–850 CrossRef CAS PubMed.
- C. J. Chang, Y. Y. Chen, C. C. Lu, C. S. Lin, J. Martel, S. H. Tsai, Y. F. Ko, D. M. Ojcius, J. D. Young and H. C. Lai, Innate Immun., 2014, 20, 301–311 CrossRef PubMed.
- S. Baby, A. J. Johnson and B. Govindan, Phytochemistry, 2015, 114, 66–101 CrossRef CAS PubMed.
- J. L. Ríos, Revista de Fitoterapia, 2008, 8, 135–146 Search PubMed.
- N. Johnston, Drug Discovery Today, 2005, 10, 1584 CrossRef PubMed.
- J. Liu, K. Kurashiki, K. Shimizu and R. Kondo, Bioorg. Med. Chem., 2006, 14, 8654–8660 CrossRef CAS PubMed.
- S. Dudhgaonkar, A. Thyagarajan and D. Sliva, Biochem. Biophys. Res. Commun., 2005, 330, 46–52 CrossRef PubMed.
- K. H. Gan, Y. F. Fann, S. H. Hsu, K. W. Kuo and C. N. Lin, J. Nat. Prod., 1998, 61, 485–487 CrossRef CAS PubMed.
- H. J. Su, Y. F. Fann, M. I. Chung, S. J. Won and C. N. Lin, J. Nat. Prod., 2000, 63, 514–516 CrossRef CAS.
- A. G. Gonzalez, F. Leon, A. Rivera, J. I. Padron, J. Gonzalez-Plata, J. C. Zuluaga, J. Quintana, F. Estevez and J. Bermejo, J. Nat. Prod., 2002, 65, 417–421 CrossRef CAS.
- X. R. Peng, J. Q. Liu, C. F. Wang, X. Y. Li, Y. Shu, L. Zhou and M. H. Qiu, J. Nat. Prod., 2014, 77, 737–743 CrossRef CAS PubMed.
- X. R. Peng, J. Q. Liu, L. S. Wan, X. N. Li, Y. X. Yan and M. H. Qiu, Org. Lett., 2014, 16, 5262–5265 CrossRef PubMed.
- X. R. Peng, J. Q. Liu, C. F. Wang, Z. H. Han, Y. Shu, X. Y. Li, L. Zhou and M. H. Qiu, Food Chem., 2015, 171, 251–257 CrossRef CAS PubMed.
- M. Dou, L. Di, L. L. Zhou, Y. M. Yan, X. L. Wang, F. J. Zhou, Z. L. Yang, R. T. Li, F. F. Hou and Y. X. Cheng, Org. Lett., 2014, 16, 6064–6067 CrossRef CAS PubMed.
- F. J. Zhou, Y. Nian, Y. M. Yan, Y. Gong, Q. Luo, Y. Zhang, B. Hou, Z. L. Zou, S. M. Wang, H. H. Jiang, J. Yang and Y. X. Cheng, Org. Lett., 2015, 17, 3082–3085 CrossRef CAS PubMed.
- X. M. Niu, M. H. Qiu, Z. R. Li, Y. Lu, P. Cao and Q. T. Zheng, Tetrahedron Lett., 2004, 45, 2989–2993 CrossRef CAS.
- R. Tanaka, M. Toyoshima and T. Yamada, Phytochem. Lett., 2011, 4, 328–332 CrossRef CAS.
- T. Nakata, T. Yamada, S. Taji, H. Ohishi, S. Wada, H. Tokuda, K. Sakuma and R. Tanaka, Bioorg. Med. Chem., 2007, 15, 257–264 CrossRef CAS PubMed.
- M. Arisawa, A. Fujita, M. Saga, H. Fukumura, M. Hayashi, M. Shimizu and N. Morita, J. Nat. Prod., 1986, 49, 621–625 CrossRef CAS.
- B. Nyasse, I. Ngantchou, J. J. Nono and B. Schneider, Nat. Prod. Res., 2006, 20, 391–397 CrossRef CAS PubMed.
- M. Hirotani, C. Ino, T. Furuya and M. Shiro, Chem. Pharm. Bull., 1986, 34, 2282–2285 CrossRef CAS.
- L. J. Lin, M. S. Shiao and S. F. Yeh, J. Nat. Prod., 1988, 51, 918–924 CrossRef CAS PubMed.
- J. Liu, K. Shimizu and R. Kondo, Fitoterapia, 2010, 81, 1067–1072 CrossRef CAS PubMed.
- J. H. Jiang, A. Jedinak and D. Sliva, Biochem. Biophys. Res. Commun., 2011, 415, 325–329 CrossRef CAS PubMed.
- N. H. Chen and J. J. Zhong, Phytomedicine, 2011, 18, 719–725 CrossRef CAS PubMed.
- W. J. Zhang, J. Y. Tao, X. P. Yang, Z. L. Yang, L. Zhang, H. S. Liu, K. L. Wu and J. G. Wu, Biochem. Biophys. Res. Commun., 2014, 449, 307–312 CrossRef CAS PubMed.
- E. F. A. Smania, F. D. Monache, A. Smania Jr, R. A. Yunes and R. S. Cuneo, Fitoterapia, 2003, 74, 375–377 CrossRef CAS PubMed.
- M. Isaka, P. Chinthanom, S. Kongthong, K. Srichomthong and R. Choeyklin, Phytochemistry, 2013, 87, 133–139 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- Y. F. Yang, J. Q. Liu, Z. R. Li, Y. Li and M. H. Qiu, Fitoterapia, 2013, 89, 278–284 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 963836 and 1405439. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra16796e |
| This journal is © The Royal Society of Chemistry 2015 |