
Significance enhancement in the conductivity of core shell nanocomposite electrolytes
Asia Rafiqueab,
Rizwan Raza*ac,
Nadeem Akrama,
M. Kaleem Ullaha,
Amjad Aliad,
Muneeb Irshade,
Khurram Siraje,
M. Ajmal Khana,
Bin Zhu*cf and
Richard Dawsong
aDepartment of Physics, COMSATS Institute of Information Technology, Lahore 54000, Pakistan. E-mail: razahussaini786@gmail.com
bHigher Education Department, Govt of Punjab, Pakistan
cDepartment of Energy Technology, Royal Institute of Technology (KTH), 10044, Stockholm, Sweden. E-mail: binzhu@kth.se
dDepartment of Physics, University of Education, Okara Campus, Lahore, 56300, Pakistan
eDepartment of Physics, University of Engineering and Technology, Lahore 54890, Pakistan
fHubei Collaborative Innovation Center for Advanced Materials, Faculty of Physics and Electronic Technology, Hubei University, Wuhan, Hubei 430062, P. R. China
gEngineering Department, Faculty of Science and Technology, Lancaster University, Lancaster, LA1 4YW, UK
First published on 6th October 2015
Abstract
Today, there is great demand of electrolytes with high ionic conductivities at low operating temperatures for solid-oxide fuel cells. Therefore, a co-doped technique was used to synthesize a highly ionically conductive two phase nanocomposite electrolyte Sr/Sm–ceria–carbonate by a co-precipitation method. A significant increase in conductivity was measured in this co-doped Sr/Sm–ceria–carbonate electrolyte at 550 °C as compared to the more commonly studied samarium doped ceria. The fuel cell power density was 900 mW cm−2 at low temperature (400–580 °C). The composite electrolyte was found to have homogenous morphology with a core–shell structure using SEM and TEM. The two phase core–shell structure was confirmed using XRD analysis. The crystallite size was found to be 30–60 nm and is in good agreement with the SEM analysis. The thermal analysis was determined with DSC. The enhancement in conductivity is due to two effects; co-doping of Sr in samarium doped ceria and it's composite with carbonate which is responsible for the core–shell structure. This co-doped approach with the second phase gives promise in addressing the challenge to lower the operating temperature of solid oxide fuel cells (SOFC).
Introduction
Solid oxide fuel cells (SOFCs) are achieving significant attention for power generation due to certain attractive features including fuel flexibility, high efficiency and potentially long operating lives.1 However, the high operating temperature range of traditional SOFCs (>900 °C) puts many constraints on system design and is particularly challenging for the choice of materials.2 Therefore, substantial efforts are in progress to lower the operating temperature regime, known as intermediate temperature (IT) range (500–750 °C).3 Lowering the operating temperatures however leads to decreased performance (lesser oxygen ion conductivity). One approach to maintain the higher conductivity is to reduce the thickness of electrolyte to minimize its ohmic resistance, but it has experienced certain limitations including a threshold thickness after which the resistance does not decrease.4 The other approach is to explore and develop new electrolyte materials possessing high ionic conductivity, at low temperatures.4Many oxygen ion conductors have been extensively studied as an electrolyte in SOFCs,5–8 and based on various families ceramic crystal structures, such as perovskites, fluorites, apatite, pyrochlore, melilite, brownmillerite, BIMEVOX, LAMOX.9–13 However, the fluorite structured materials i.e. ceria (CeO2), zirconia (ZrO2) and Bi2O3 are considered as ideal oxide ion conductors. Ideal fluorite structure is a face centered cubic (FCC) array of cations, while anions sit on the tetrahedral sites, where the conduction mechanism occurs in the anion sub-lattices by means of ‘vacancy migration’. Only ceria (CeO2) retains its fluorite structure at room temperature, while both ZrO2 and Bi2O3 have monoclinic structures which can be stabilized with other cation like yttria.
At intermediate temperature range, the performance of SOFC is largely dominated by two processes which are; the oxygen reduction reaction (ORR) that occurs at the cathode and the ionic conductivity of the electrolyte.5 Yttria stabilized zirconia (YSZ) as an electrolyte is a good choice and is reliable due to its structural and thermodynamic stability. But it possess low ionic conductivity at IT range although a conductivity of 4.2 × 10−2 S cm−1 at temperature 800 °C has been reported.6 Also Bi2O3 showed highest conductivity of 1 S cm−1 keeping its fluorite structure but within a very narrow temperature range, 730–804 °C.14,15 These structural changes be avoided by appropriate doping for example; for Bi1.6Er0.4O3, 2 × 10−2 S cm−1 conductivity is reported at 500 °C, while high conductivities of 10−3 to 10−2 S cm−1 has been achieved for Bi12.5La1.5ReO24.5 and Bi0.85Pr0.105V0.045O1.545 at temperatures 300–400 °C.9,16,17
Among these fluorite structured materials, ceria (CeO2) based materials are attracting a great amount of interest as electrolytes for SOFC due to their high conductivity at lower temperatures and good stability. In reducing atmosphere, ceria doped with rare earth elements is partially reduced and exhibits electronic conductivity,18 with typical peak conductivity (∼0.01 S cm−1) reported at 500 °C Ce0.8Sm0.2O1.9 and Ce0.9Gd0.1O1.95,19 and is potentially a significant problem which must be mitigate in any practical device. In ceria, the ionic conductivity is related to the formation of oxygen vacancy and its migration,20,21 and these vacancies are produced for compensating dopant cations. Though pure ceria is a poor oxide-ion conductor as low conductivity of ∼10−5 S cm−1 doping results in an increased concentration of oxygen vacancies according to the reaction.2
 | (1) |
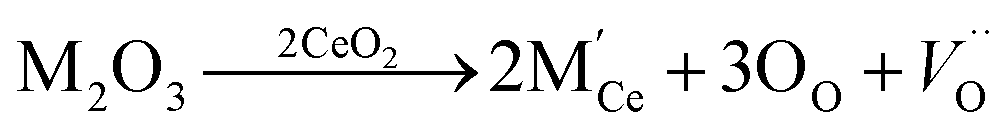 | (2) |
The oxygen vacancy in doped ceria depends on the nature of the dopant and its amount.24,25 Its ionic conductivity is also affected by the ‘lattice strain’ as generated due to the ionic-radius mismatch between the dopant and host ions.3 Therefore, it is very important to have an appropriate choice of dopant and its amount to minimize the lattice strain and consequently to enhance the ionic conductivity.26–29 It has been reported that sufficient oxygen vacancies are produced by doping of 20 mol% divalent or trivalent cation in ceria,30 for example samarium doped ceria (SDC) and gadolinium doped ceria (GDC).26,31 But there is deterioration of the ionic conductivity of CeO2 due to the clustering of oxygen vacancy (or defect association) as the trivalent dopant (M3+) content increases above 20% mol, which leads to the generation of few mobile vacancies.32
For ceria doped with different cations, only samaria doped ceria (SDC) has showed highest conductivity as reported by Eguchi et al.,33 as the ionic radius of samaria (1.079 Å) matches with ceria (0.94 Å). Although, it is considered that minimum difference of ‘ionic radii mismatch’ between dopant and host results in the increase of conductivity.34,35 But it does not seem always acceptable because the ionic radius of yttria (1.019 Å) is closer to ceria as compared to samaria.36 Therefore, it is still debatable whether only the ionic radius of the dopant determines the oxide ion conductivity or some other parameters also plays an important role.
However, are cognized route for improving the ionic conductivity of such fluorite structured materials is co-doping (also known as doubly doped) i.e. doping with two or more than two differentiation species.5 This approach was first employed by Politova and Irvine,37 for solid electrolytes, when they studied the doping of scandia (scandium) and yttria (yttrium) with zirconia. They reported that only a small content of yttria (yttrium) is necessary for stabilizing the cubic fluorite-structure, as further addition of it to Sc-doped ZrO2 decreases the conductivity of material.37 However, contrary to the role of co-doping in ZrO2 for stabilizing its structure, co-doping (it) can be used in CeO2 for increasing its ionic conductivity by reproducing the ionic radius of the ideal dopant or the lattice constant. The purpose of the doping is to achieve an effective or average cation radius, very close to ceria; hence to minimize the ‘average strain’ as developed by dopant cations.5 Co-doping effects of different trivalent metals like Y, Sm, Nd, Pr, La on gadolinia doped ceria (GDC) were studied by Kim et al.,38 and found an increase in ionic conductivity with Sm co-doping. Yamamura et al.,39 discussed the effect of co-doping on the system Ce1−x−yLaxMyO2−δ with M = Ca or Sr. For singly (Ce0.8Ln0.2O1.9, where Ln = Y, Sm, Nd, La) and doubly doped ceria (Ce0.8La0.1Y0.1O1.9), the ionic conductivity was investigated by Yoshida et al.,40 using extended X-ray absorption fine structure (EXAFS). Andersson et al. calculated theoretically using DFT the effect of co-doping in ceria with Nd/Sm and Pr/Gd and predicted that it can enhance the ionic conductivity as compared to singly doped ceria.41 Omar et al.,42 reported an increase in ionic conductivity (0.014 S cm−1 at 550 °C) with Ce0.85Nd0.075Sm0.075O1.925, based on these theoretical backgrounds,41 which was 30% as compared to Ce0.9Gd0.1O1.95. Similar results were reported by Ramesh et al.,43 for co-doping of Gd and Pr with ceria and found 11.5% higher ionic conductivity than GDC. Sha et al.,44 studied the effect of co-doping of La and Y with ceria and found an improvement in the ionic conductivity. Yeh and Chou,30 investigated co-doping of strontium (1.25 Å) with SDC and achieved good conductivity of 0.061 S cm−1 with Ce0.78Sm0.2Sr0.02O1.88 at 800 °C which was twice of singly doped SDC. Recently, Gao et al.,45 reported an increase in the bulk conductivity for Ce0.8(Sm0.7Sr0.3)0.2O2−δ.
The co-doping is a good approach for the structure modification of ceria-based material to improve the ionic oxide conductivity at low temperature range (300–600 °C). Sr is very attractive due to its distinctive co-doping effect into the ceria host lattice and the ionic-radius compatibility with the host cation.19 The introduction of the co-doped ceria with Sr can lead to the improved ionic conductivity.30,46 The ionic conductivity of co-doped ceria can be further increased by introducing carbonates as a second phase. The carbonate phase as a core–shell structure provides interface for ion conduction and is reported in our previous work.47–49
In this paper, we report first time on co-doping of carbonate based ceria composite with different concentrations/compositions of Sr and Sm for electrolytes suitable for IT-SOFC. The crystal structure and surface morphology of the synthesized electrolyte materials was studied by X-ray diffraction (XRD), scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX). The existence of second phase responsible for core–shell structure is confirmed by TEM analysis. We measured an increased ionic conductivity at low temperature range and consequently the enhanced performance of the fuel cell.
Experimental
Sample/electrolyte preparation
Sr & Sm co-doped ceria core with Na2CO3 carbonate shell is named as nanocomposite electrolytes were synthesized with different compositions Sr0.2Sm0.0Ce0.8O2−δ–carbonate (sample-1), Sr0.2Sm0.1Ce0.7O2−δ–carbonate (sample-2), Sr0.1Sm0.2Ce0.7O2−δ–carbonate (sample-3) and Sr0.1Sm0.1Ce0.8O2−δ–carbonate (sample-4) by a co-precipitation technique.47,50 Stoichiometric amounts of cerium nitrate hexahydrate Ce(NO3)3·6H2O (Sigma Aldrich 99%, USA), samarium nitrate hexahydrate Sm(NO3)3·6H2O (Sigma Aldrich 99%, USA) and strontium nitrate Sr(NO3)2 (Sigma Aldrich 99%, USA) were mixed and dissolved in de-ionized water to make a 0.1 M solution. The nitrate solution was stirred for 2 hours and 0.2 M Na2CO3 solution was prepared and added drop wise in the nitrate solution. The resulting precipitate was rinsed with de-ionized water and then dried in oven at 250 °C for 2 hours. The dried powder was subsequently calcined 850 °C in furnace for 4 hours. The same procedure was used to prepare all the samples.The solid state reaction (SSR) method was used to prepare the Li–Ni–Cu–Zn (LNCZ) oxide electrodes. Li2CO3·3H2O (Sigma Aldrich, 99% USA), Ni2(CO3)3·6H2O (Sigma Aldrich, 99% USA),Cu(CO3)3 (Sigma Aldrich, 99% USA) and Zn(NO3)2 (Sigma Aldrich, 99% USA) were mixed in a weight ratio of 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:2.5:7. These were grinded in mortar pestle and then calcined for 4 hours at 800 °C. Nickel and copper oxides were used as a catalyst in the anode.
:7:2.5:7. These were grinded in mortar pestle and then calcined for 4 hours at 800 °C. Nickel and copper oxides were used as a catalyst in the anode.
Characterization
The calcined powdered samples were characterized by X-ray diffraction analysis (XRD) using X-ray diffractometer (PANalytical X'Pert Pro MPD, Phillips, Netherlands) with monochromated Cu Kα radiation (λ = 0.15418 nm). The lattice constant and lattice parameter of the materials were determined from the XRD peaks. The average crystallite size D was determined by using the Scherrer's equation:|
D = (0.9λ)/(βcosθ)
| (3) |
The microstructure and morphology of the samples were examined using scanning electron microscope (FE-SEM, Carl Zeiss, Germany). In order to confirm the second phase (core–shell) and microstructure analysis TEM was performed on a JEOL NM-200 and operated at 200 kV. The thermal behaviors of the composite electrolytes were investigated by thermogravimetry analysis (TGA) (model Q600, USA), and the samples were heated from 25 °C to 1000 °C at a rate of 10 °C min−1. The thermal expansion co-efficient and change in volume of solids were measured using a NETZSCH model 402 C pushrod dilatometer. This dilatometer was equipped with a SiC furnace capable of operation between room temperature and 1600 °C. The system is vacuum tight, allowing measurements to be carried out in pure inert or oxidizing atmospheres, as well as under vacuum.
Pellet fabrication for conductivity and performance measurement
For conductivity measurements, 1.15 mm thick pellets with 13 mm diameter having an active area of 0.84 cm2 were made with the different compositions of the electrolyte under 40 MPa pressure and sintered in air at 700 °C for 60 minutes. To collect/measure the current, silver paste was used on both sides of the pellet and dried at 600 °C for 30 min. The conductivity of the sintered samples was measured by four point dc probe method at (300–600 °C) temperature range in air. The distance between voltage probe and current probe was kept as 1.2 mm. The Probe station (KeithLink, China) with 4 probes of tungsten integrated with DC current source 2450 (Keithley Instruments, USA) were used for current and voltage measurements. The conductivity was calculated using the collected data from KickStart software (Keithley instruments, USA), with the following formula;| σ = L/RA | (4) |
For fuel cell pellets fabrication, same procedure was used to make the cells (LNCZ + Sr–SDC|Sr–SDC|LNCZ + Sr–SDC). Fuel cell was 13 mm in diameter and 0.8 mm in thickness (anode thickness 0.30 mm, electrolyte 0.30 mm and cathode 0.20 mm), so the cell has an electrolyte supported configuration. The cell performance was measured with a computerized instrument (Fuel Cell Electronic load, Model: IT8511, China) at 600 °C and H2 gas was used as a fuel with a flow rate of 100 ml min−1 at atmospheric pressure and ambient air was used as an oxidant.
Results and discussion
Phase analysis and microstructure
Fig. 1 displays the pattern of XRD for co-doped Sr/Sm ceria–carbonate electrolyte with different compositions which are indexed by using the Mjad-5 software. The indexed patterns are showed that all the compositions have a single phase of CeO2 oxide, and clearly seen that both Sr and Sm have been doped properly in ceria. It also shows that all co-doped ceria electrolytes have cubic fluorite structure (JCPDS Card no. 39-0394). The values of angle 2θ of doped ceria shift slightly with change in the composition of Sr and Sm. The average crystallite size of each composition was calculated form peak data of ceria phase by using the Scherrer's equation and is shown in the Table 1. The results showed that the composition Sr0.1Sm0.1Ce0.8O2−δ–carbonate has the smallest average crystallite size 29.637 nm. It can also be seen that lattice parameter of ceria oxide are varied with the ratios of Sr and Sm contents. This lattice parameter of ceria oxide are changed due to due to slightly difference of ionic radii of Ce4+ (0.94 Å) with Sm3+ (1.079 Å) and Sr2+ (1.25 Å). These values are in complete agreement with Vegord's rule and also verify that the prepared samples of doped ceria are indeed ceria based solid solution.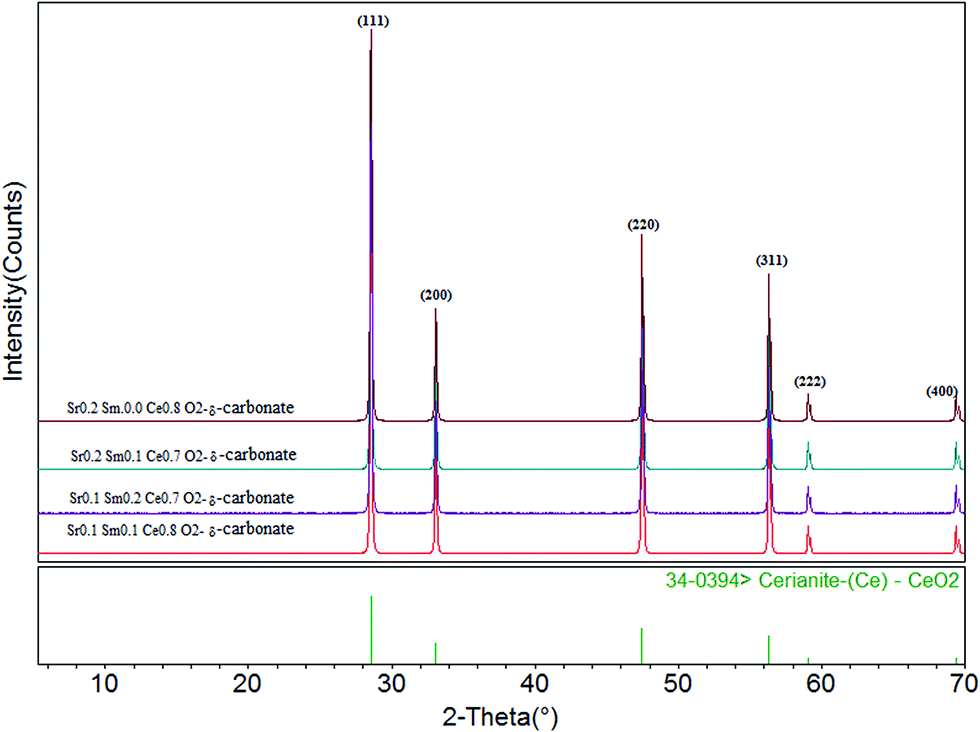 | ||
| Fig. 1 X-ray diffraction pattern for different compositions of Sr/Sm ceria–carbonate electrolytes. | ||
| Sr. No. | Composition | Crystallite size of sintered powder (nm) | Lattice parameter (Å) |
|---|---|---|---|
| 1 | Sr0.2Sm0.0Ce0.8O2−δ–carbonate | 47.898 | 5.4037 |
| 2 | Sr0.2Sm0.1Ce0.7O2−δ–carbonate | 43.239 | 5.4186 |
| 3 | Sr0.1Sm0.2Ce0.7O2−δ–carbonate | 31.276 | 5.4227 |
| 4 | Sr0.1Sm0.1Ce0.8O2−δ–carbonate | 29.637 | 5.4225 |
The microstructural morphologies for all compositions of as prepared powder/electrolytes were studied using SEM. Fig. 2(a) shows the typical microstructure of Sr0.1Sm0.1Ce0.8O2−δ–carbonate and indicating that particles are homogeneous and distributed uniformly. In Fig. 2(a), it can also be observed that the prepared electrolyte is not porous and is quite dense. The particle shapes are irregular and the average size is ∼30–60 nm, which agrees adequately with the XRD analysis.
 | ||
| Fig. 2 (a) SEM micrograph for Sr0.1Sm0.1Ce0.8O2−δ–carbonate with closed view of core shell particles (b) EDX spectrum of co-doped ceria with Sr/Sm–carbonate (c) TEM micrograph for Sr0.1Sm0.1Ce0.8O2−δ–carbonate core shell. | ||
Micrograph of Fig. 2(a) clearly represents the carbonate phase or second phase with the ceria phase, as there is a distinct contrast between the inner and outer shell of the particles. This indicates the presence of core–shell structure as the percolation of amorphous carbonates is obvious. Such two phase regions facilitate the ionic conduction by constructing ion conducting paths.48 In order to confirm the carbonate phase and contents in the electrolyte, EDX spectrum was also performed and Fig. 2(b) shows the formation of Na2CO3-core shell on Sr–SDC. The amorphous nature of the shell can also be verified from the XRD pattern which shows no peak of Na2CO3. This also compliments the SEM analysis which reveals the shallow layer of Na2CO3 on the particles.
The high-resolution TEM image of a small part of co-doped ceria shows the crystal structure and particle size in Fig. 2(c). The presence of secondary phase and core shell was observed and shown in Fig. 2(c). It can be clearly seen that shell layer is very thin and, several nm in thickness, was formed outside the co-doped Sr–SDC particle.
The formation of core shell layer will act as a barrier to electronic conduction between anode and electrolyte. This shell will protect SDC from partial reduction by the fuel thus further reducing any electronic current.
Thermal analysis
Fig. 3(a) and (b) describe the temperature-dependent mass variations and heat flow rate of the ceria co-doped nanocomposite electrolyte in air and reducing varigon atmospheres at room temperature to 650 °C. In the temperature range of 25 °C to 150 °C, a mass loss step of 2.9% was observed in varigon and air environments due to the evaporation of absorbed moisture/water, which was escorted by an endothermic DSC peak with an enthalpy of ∼66 J g−1. In the next step of temperature range 150 °C to 400 °C, further mass loss of 0.4–0.5% occurred, which could be due to the burn-up of impurities in ceria co-doped nanocomposite electrolyte. Above the temperature of 400 °C, there was no mass loss to observe in the air atmosphere, but in the varigon environment showed 0.5% mass loss which is due to reduction environs. A small endothermic DSC peak was observed around the temperature of 600 °C in both air and reducing varigon atmospheres, where the phase transformation may be occurred.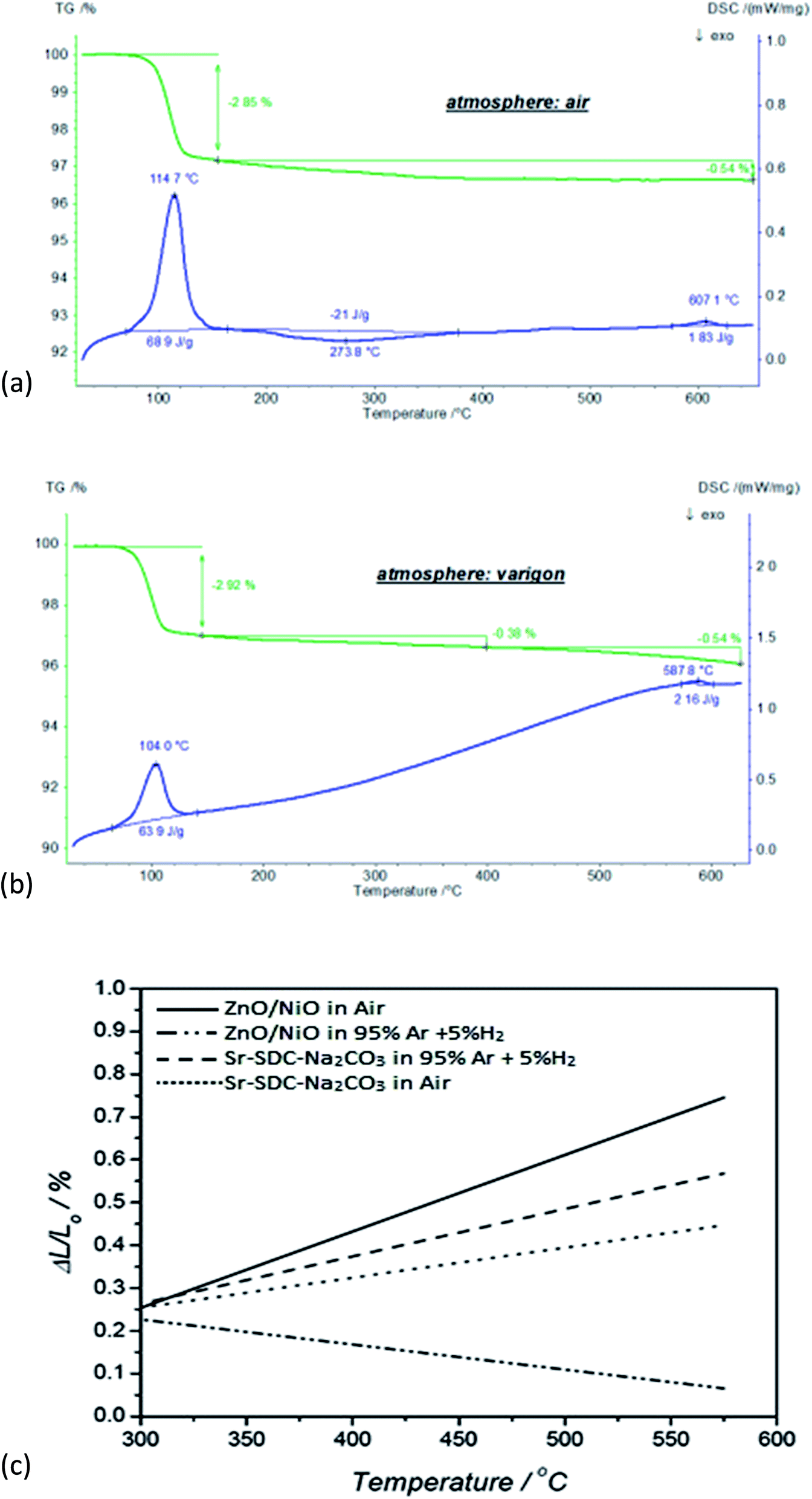 | ||
| Fig. 3 (a) TGA/DSC of Sr/Sm ceria–carbonate electrolytes with a ramp rate of 10 °C min−1 in H2 atmosphere, (b) TGA/DSC of Sr/Sm ceria–carbonate electrolytes with a ramp rate of 10 °C min−1 in argon atmosphere, (c) thermal expansion of ZnO/NiO and Sr/SDC–carbonate both in air and H2 (diluted with argon), in which ΔL/L is the relative variation of the length of the samples. | ||
In order to evaluate the mechanical compatibility of the ZnO/NiO materials with the electrolyte, thermal expansion measurements were performed. Fig. 3(b) illustrates the variations of the ΔL/L (coefficient of linear expansion) values in the range of 300–550 °C. It shows good match of the ZnO/NiO materials and Sr/Sm ceria–carbonate in the air. In the H2 atmosphere, it shows a shrinkage of ZnO/NiO materials due to the Ni–Zn phase formation with ZnO/NiO reduced by H2 and succedent Ni–Zn transformation around 530 °C. In the application of low temperature (LT) SOFCs stack using ZnO/NiO electrodes, it should be operated lower than 530 °C in order to prevent the mechanical degradation of the electrode. It can been seen from the Fig. 3(b) that with increasing temperature, the difference between the curves of samples exposed to different ambient conditions also increases. In the case of Sr doped samples the difference between the curves is not very significant as compared to the ZnO/NiO for different ambient environment due to fact that degradation in ZnO/NiO samples starts at higher temperatures.
Conductivity and cell performance
As discussed earlier, there are two major challenges for the commercialization of SOFC, one is to lower the operating temperature and second is to explore new, more cost-effective, and stable materials/compositions. Here the Sr-divalent cation has been used as a co-dopant due to its low cost and easy availability.51 From Fig. 4(a), it can be seen that Sr/Sm ceria–carbonate system shows higher ionic conductivity at low temperature as compare to singly doped ceria with same conductivity at 1000 °C. Furthermore, the co-doing of Sr in SDC and carbonate as core shell could help to overcome the electronic conduction of CeO2 in anodic environment and also to enhance the density of solid electrolyte.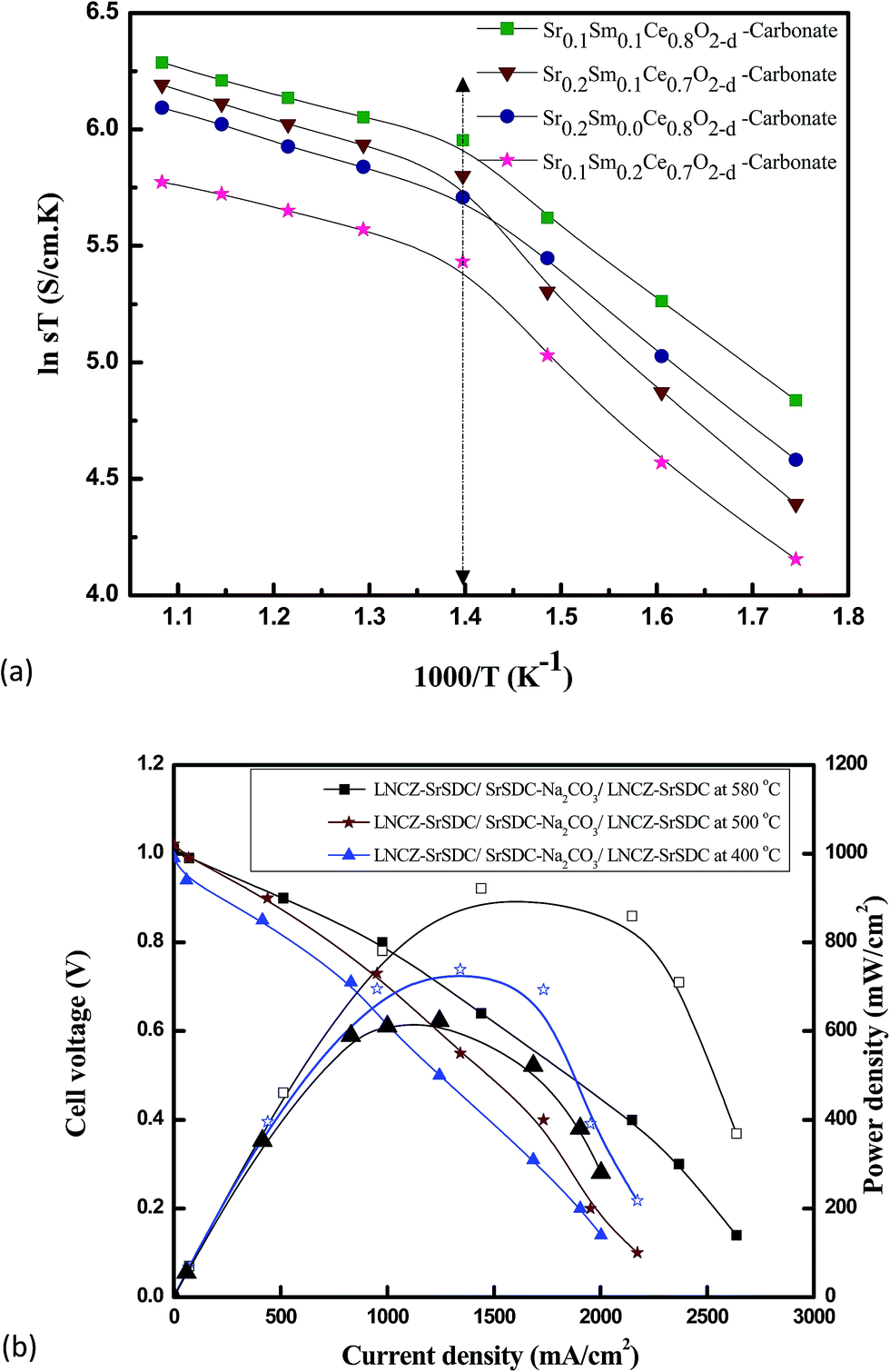 | ||
| Fig. 4 (a) Arrhenius plot for different Sr/Sm ceria–carbonate electrolytes in air (b) I–V/I–P characteristics of a fuel cell at different temperatures 400 °C, 500 °C, 580 °C. | ||
The main purpose of adding the Na2CO3 in electrolyte is to create a second phase as a core shell, which also reported previously in our work.48,49 This may form a large interface region for ion conduction paths between the SDC and the carbonate at elevated temperatures to greatly enhance the material conductivity.48,49 This interface has, in principle, no bulk structural limit for the creation of high concentration of mobile ions, and can thus be greatly disordered. This implies that such interfaces have the capacity to contain higher mobile ion concentration than that of the bulk. The electric field distribution in the interfaces between two phases is the key to realizing the interfacial super-ionic conduction, allowing ions to move on particle's surfaces or interfaces by high conductivity pathways.
The higher conductivity of the prepared composite materials at lower temperature is also due to the amorphous nature of Na2CO3 shell. It can protect the active surface of SDC and interfaces in nanoscale to enhance the nano-material stability as well as further promote the oxygen ion transportation through the interfacial mechanism.47 The use of core–shell co-doped ceria–carbonate nanocomposite electrolytes resulted in a greater conductivity and thermal stability as compared to that of single-phase ceria, and a high ionic conductivity in excess of 0.5 S cm−1 at 300–600 °C.
Arrhenius plot was drawn from the total ionic conductivity data by curve fitting to calculate the activation energies (Ea) of the Sr0.1Sm0.1Ce0.8O2−δ–carbonate Sr0.2Sm0.0Ce0.8O2−δ–carbonate, Sr0.2Sm0.1Ce0.7O2−δ–carbonate, and Sr0.1Sm0.2Ce0.7O2−δ–carbonate, nano-composite electrolytes under air atmosphere in the temperature range 300–650 °C and results is shown in Fig. 4(a). It can be seen clearly from the Fig. 4(a) that Sr0.1Sm0.1Ce0.8O2−δ–carbonate exhibits high ionic conductivity as compared to the others. Its conductivities increase due to increase of oxygen ions transportation from created large number of oxygen vacancies at high temperature. The doping of strontium in SDC significantly alters its ionic conductivity as reported earlier e.g. T. H. Yeh reported 0.061 S cm−1 at 800 °C, N. Jaiswal reported 0.004 S cm−1 at 500 °C and many others reported.30,42,45,52,53 The change in the ionic conductivity due to the doping of strontium can be related to decrease in lattice binding energy that result into increased numbers of oxygen vacancies. The number of oxygen vacancies is directly related to the conductivity of the material.30,55 At lower temperature, it has may be less lattice binding energy and defects in the interface phases are not highly mobility for the oxygen ions.
It can also be seen that there is sharply jump around 400 °C, where it could be related to glass transition temperature.49 The behaviors of conductivities in the air atmosphere was increased with increased the temperature. The enhanced ionic conductivity of co-doped ceria due to strontium doping can be attributed to; (i) maximized non interfering oxygen vacancies, (ii) the average radii of co-doping divalent cations close to that of Ce4+ and (iii) small average binding energy. The Table 2 depicts activation energies due to oxygen ions migration for the prepared samples calculated from the Arrhenius equation.
| Composition | Activation energy (eV) 300–650 °C |
|---|---|
| Sr0.2Sm0.0Ce0.8O2−δ–carbonate | 0.23 |
| Sr0.2Sm0.1Ce0.7O2−δ–carbonate | 0.24 |
| Sr0.1Sm0.2Ce0.7O2−δ–carbonate | 0.21 |
| Sr0.1Sm0.1Ce0.8O2−δ–carbonate | 0.20 |
The ionic transference number (τion) of ceria co-doped electrolyte was obtained by Hebb–Wagner's DC polarization method54,55 at 600 °C. The following equation was used for calculation.
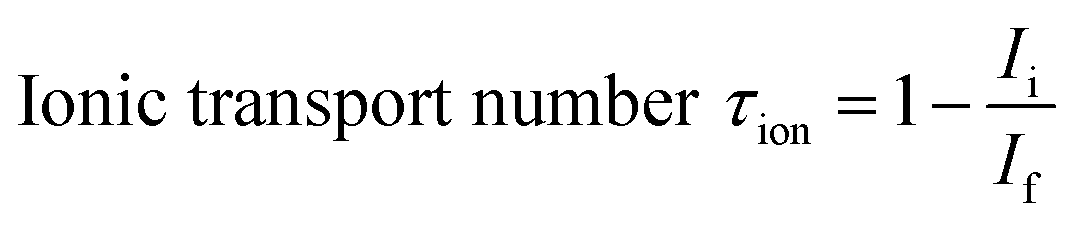 | (5) |
The ionic transference numbers (τi) of sample Sr0.1Sm0.1Ce0.8O2−δ–carbonate as calculated using dc polarization technique was found to be 0.90.
The characteristics curves of I–V/I–R for different temperatures 400 °C, 500 °C, 580 °C are represented in Fig. 4(b) exhibiting that maximum power density of 900 mW cm−2 is achieved at 580 °C. Open circuit voltage (OCV) and current data were recorded for cells (symmetric) at temperature range 400−580 °C, using the Sr0.1Sm0.1Ce0.8O2−δ–carbonate, as electrolyte and LNCZ–SrSDC as electrodes. This sample was used because Sr0.1Sm0.1Ce0.8O2−δ–carbonate has the maximum conductivity.
Wenquan et al.,56 achieved a power density of 190 mW cm−1 at 800 °C for the electrolyte La0.9Sr0.1Ga0.8Mg0.2O3 (LSGM) and another research group,56 reported an increase in the power density of 170 mW cm−1 at 1073 K with SDC electrolyte and Sr-doped samarium cobaltite cathode with the addition of RuO2. However, no report has been seen previously for the calculation of power density of Sr–SDC as an electrolyte. In this present research, a maximum performance has been achieved at low temperature 400–580 °C.
The reduced IR drop from electrolyte ohmic behavior can account for the higher performance at such low temperature. The two phase electrolyte produced by the ceria/carbonate composite, as found, displays a higher ionic conductivity possibly due to enhanced ionic conduction pathways and which makes for the excellent performance demonstrated.
Conclusions
The results presented in this article show the significant enhancement in the ionic conductivity of the electrolyte which can improve fuel cell performance at lower temperatures. Excellent performance was obtained at 550 °C where a maximum power density of 900 mW cm−2 was measured running on excess pure hydrogen at modest pressures and ambient. The ionic conductivity of the best composition of co-doped composite electrolyte was 0.5 S cm−1. The enhanced conductivity is likely to be attributed to the effect of co-doping and of carbonate phases, which leads to a higher ionic conductivity pathways in the electrolyte.This research provides fundamental studies about co-doped ionic composite conductors which can lead to lower the operating temperature of SOFC. The results using these materials can strongly support the development of the low temperature SOFC for commercialization with an ultra-low cost and reliable performance. At present, the development of composite co-doped electrolyte materials and its application in LT-SOFCs are still at an initial stage. Development of SOFC technology operating at 300–600 °C also opens up new market opportunities.
Acknowledgements
Authors acknowledge Indigenous scholarship, Higher Education Commission (HEC), the start-up research grant from Higher Education Commission (HEC), Pakistan and COMSATS Research Grant Program (CRGP) to support this work.References
- N. Q. Minh and T. Takahashi, Science and Technology of Ceramic Fuel Cells, Elsevier, Amsterdam, 1995 Search PubMed.
- O. Parkash, N. Singh, N. K. Singh and D. Kumar, Solid State Ionics, 2012, 212, 100–105 CrossRef CAS PubMed.
- D. J. L. Brett, A. Atkinson, N. P. Brandon and S. J. Skinner, Chem. Soc. Rev., 2008, 37, 1568–1578 RSC.
- D. S. Lee, W. S. Kim, S. H. Choi, J. Kim, H. W. Lee and J. H. Lee, Solid State Ionics, 2005, 176, 33–39 CrossRef CAS PubMed.
- M. Burbano, S. Nadin, D. Marrocchelli, M. Salanne and G. W. Watson, Phys. Chem. Chem. Phys., 2014, 16, 8320 RSC.
- V. V. Kharton, E. N. Naumovich and A. A. Vecher, J. Solid State Electrochem., 1999, 3, 61–81 CrossRef CAS.
- S. Omar, E. D. Wachsman and J. C. Nino, Solid State Ionics, 2006, 177, 3199 CrossRef CAS PubMed.
- R. M. Ormerod, Chem. Soc. Rev., 2003, 32, 17–28 RSC.
- V. V. Kharton, F. M. B. Marques and A. Atkinson, Solid State Ionics, 2004, 174, 135–149 CrossRef CAS PubMed.
- P. Lacorre, F. Goutenoire, O. Bohnke, R. Retoux and Y. Laligant, Nature, 2000, 404, 856–858 CrossRef CAS PubMed.
- E. Kendrick, M. S. Islam and P. R. Slater, J. Mater. Chem., 2007, 17, 3104–3111 RSC.
- J. C. Boivin and G. Mairesse, Chem. Mater., 1998, 10, 2870–2888 CrossRef CAS.
- X. Kuang, M. A. Green, H. Niu, P. Zajdel, C. Dickinson, J. B. Claridge, L. Jantsky and M. J. Rosseinsky, Nat. Mater., 2008, 7, 498–504 CrossRef CAS PubMed.
- H. A. Harwig and A. G. Gerards, J. Solid State Chem., 1978, 26, 265–274 CrossRef CAS.
- H. A. Harwig and A. G. Gerards, Thermochim. Acta, 1979, 28, 121–131 CrossRef CAS.
- R. Punn, A. M. Feteira, D. C. Sinclair and C. Greaves, J. Am. Chem. Soc., 2006, 128, 15386–15387 CrossRef CAS PubMed.
- M. Benkaddour, S. Obbade, P. Conflant and M. Drache, J. Solid State Chem., 2002, 163, 300–307 CrossRef CAS.
- H. Inaba and H. Tagawa, Solid State Ionics, 1996, 83, 1–16 CrossRef CAS.
- B. C. H. Steele, Solid State Ionics, 2000, 129, 95–110 CrossRef CAS.
- R. Raza, G. Abbas, Y. Ma, X. Wang and B. Zhu, Solid State Ionics, 2011, 188, 58–63 CrossRef CAS PubMed.
- P. P. Dhlabhai, J. B. Adams, P. Crozier and R. Sharma, J. Chem. Phys., 2010, 132, 094104 CrossRef PubMed.
- C. Peng, Y. N. Liu and Y. X. Zheng, Mater. Chem. Phys., 2003, 82, 509 CrossRef CAS.
- P. P. Sahoo, J. L. Payne, M. Li, J. B. Claridge and M. J. Rosseinsky, J. Phys. Chem. Solids, 2015, 76, 82–87 CrossRef CAS PubMed.
- M. Mogensen, N. M. Sammers and G. A. Tompsett, Solid State Ionics, 2000, 129, 63 CrossRef CAS.
- A. Ismail, J. Hooper, J. B. Giorgi and T. K. Woo, Phys. Chem. Chem. Phys., 2011, 13, 6116 RSC.
- D. J. Kim, J. Am. Ceram. Soc., 1989, 72, 1415 CrossRef CAS PubMed.
- J. Kilner, Solid State Ionics, 1983, 8, 201 CrossRef CAS.
- Y. Y. Liu, B. Li, X. Wei and W. Pan, J. Am. Ceram. Soc., 2008, 91, 3926 CrossRef CAS PubMed.
- S. Omar, E. D. Wachsman and J. C. Nino, Solid State Ionics, 2008, 178, 1890 CrossRef CAS PubMed.
- T. H. Yeh and C. C. Chou, Phys. Scr., 2007, T129, 303–307 CrossRef CAS.
- H. Yahiro, K. Eguchi and H. Arai, Solid State Ionics, 1989, 36, 71–75 CrossRef CAS.
- C. T. Chen, S. Sen and S. Kim, Chem. Mater., 2012, 24, 3604–3609 CrossRef CAS.
- K. Eguchi, T. Setoguchi, T. Inoue and H. Arai, Solid State Ionics, 1992, 52, 165 CrossRef CAS.
- J. A. Kilner and R. J. Brook, Solid State Ionics, 1982, 6, 237 CrossRef CAS.
- C. R. A. Catlow, Solid State Ionics, 1984, 12, 67 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- T. I. Politova and J. T. S. Irvine, Solid State Ionics, 2004, 168, 153–165 CrossRef CAS PubMed.
- N. Kim, B. H. Kim and D. Lee, Electrochem. Soc., Proc., 1999, 201–208 CAS.
- H. Yamamura, E. Katoh, M. Ichikawa, K. Kakinuma, T. Mori and H. Haneda, Electrochemistry, 2000, 68, 455–459 CAS.
- H. Yoshida, H. Deguchi, K. Miura, M. Horiuchi and T. Inagaki, Solid State Ionics, 2001, 140, 191–199 CrossRef CAS.
- D. A. Andersson, S. I. Simak, N. V. Skorodumova, I. A. Abrikosov and B. Johansson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3518–3521 CrossRef CAS PubMed.
- S. Omar, E. D. Wachsman and J. C. Nino, Solid State Ionics, 2008, 178, 1890–1897 CrossRef CAS PubMed.
- S. Ramesh and K. C. J. Raju, Int. J. Hydrogen Energy, 2012, 37, 10311–10317 CrossRef CAS PubMed.
- X. Sha, Z. Lü, X. Huang, J. Miao, Z. Ding, X. Xin and W. Su, J. Alloys Compd., 2007, 428, 59–64 CrossRef CAS PubMed.
- Z. Gao, X. Liu, B. Bergman and Z. Zhao, J. Power Sources, 2012, 208, 225–231 CrossRef CAS PubMed.
- X. Sha, Z. Lü, X. Huang, J. Miao, Z. Ding, X. Xin and W. Su, J. Alloys Compd., 2007, 428, 59 CrossRef CAS PubMed.
- R. Raza, X. Wang, Y. Ma and B. Zhu, J. Power Sources, 2010, 195, 6491 CrossRef CAS PubMed.
- X. Wang, Y. Ma, R. Raza, M. Muhammed and B. Zhu, Electrochem. Commun., 2008, 10, 1617–1620 CrossRef CAS PubMed.
- Y. Ma, X. Wang, R. Raza, M. Muhammed and B. Zhu, Int. J. Hydrogen Energy, 2010, 35, 2580–2585 CrossRef CAS PubMed.
- H. M. Li, Q. C. Zhu, Y. L. Li, M. C. Gong, Y. D. Chen, J. L. Wang and Y. Q. Chen, J. Rare Earths, 2010, 28, 79 CrossRef CAS.
- B. D. Cullity, Elements of X-ray Diffraction, Addison-Wesley, Reading, MA, 1956 Search PubMed.
- B. Zhu and M. D. Mat, Int. J. Electrochem. Sci., 2006, 1, 383–402 CAS.
- N. Jaiswal, S. Upadhyay, D. Kumar and O. Parkash, J. Power Sources, 2013, 222, 230–236 CrossRef CAS PubMed.
- C. Wagner, Z. Phys. Chem., 1933, B21, 25–47 CAS.
- M. H. Hebb, J. Chem. Phys., 1952, 20, 185–190 CrossRef CAS PubMed.
- G. Wenquan, G. Srikanth and B. P. Uday, J. Power Sources, 2006, 160, 305–315 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |