
Structurally novel C17-sesquiterpene lactones from Ainsliaea pertyoides†
Zhi-Ran
Shi
ab,
Yun-Heng
Shen
*b,
Xian-Yuan
Zhang
c,
Xin
Fang
b,
Ren-Tao
Zeng
b,
Qing-Xin
Liu
ab,
Zhi-Guo
Zhuo
b,
Feng
Feng
*a and
Wei-Dong
Zhang
*abc
aDepartment of Natural Medicinal Chemistry, China Pharmaceutical University, Nanjing 210009, P. R. China. E-mail: fengsunlight@163.com; wdzhangy@hotmail.com
bDepartment of Phytochemistry, School of Pharmacy, Second Military Medical University, Shanghai 200433, P. R. China. E-mail: shenyunheng@hotmail.com
cShanghai Institute of Pharmaceutical Industry, Shanghai 200040, P. R. China
First published on 16th October 2015
Abstract
Pertyolides A (1) and B (2), two unprecedented C17-eudesmanolides with extended side chains, and one rare C17-guaianolide, pertyolide C (3), along with 15 known sesquiterpene lactones were isolated from the whole plant of Ainsliaea pertyoides. Their structures were elucidated by extensive spectroscopic analysis. The absolute configuration of compound 1 was assigned by X-ray crystallography. All isolates were evaluated for their cytotoxic activities against four human tumor cell lines A549, HCT116, MGC803, CCRF-CEM, and only alantolactone (5) showed significant inhibitions against the tumor cell lines tested.
The genus Ainsliaea (family Compositae), consisting of nearly 70 species, widely spread throughout the South East Asian region, are receiving much attention due to their remarkable biological activities and chemical diversities.1–5 As an important part of our research for screening bioactive constituents from natural sources, great efforts of our group have been devoted to the phytochemical investigations on plants of the genus Ainsliaea, which resulted in a series of biologically active sesquiterpenoid lactone dimmers and triterpenoid derivatives with new carbon frameworks, as well as other types of constituents such as triterpenes, steroids, flavonoids, etc.6–8
With the aim of discovering bioactive natural products we carried out an investigation on the chemical constituents of Ainsliaea pertyoides, one of the 48 Chinese endemic species, which is a medicinal herb growing mainly in southeast of China and only a few natural products have been discovered.9 It is an ethnomedicinal plant used in traditional Chinese medicine for the treatment of various diseases, including bone fracture and ankylosing spondylitis. From this study, we have isolated two unprecedented C17-eudesmanolides with extended side chains pertyolides A (1) and B (2), one new C17-guaianolide pertyolide C (3) (Fig. 1) and 15 known sesquiterpene lactones (Fig. S1, ESI†). It is worthwhile to mention that compounds 1 and 2 are two unusual eudesmane-type sesquiterpenoid with a C17 skeleton. Herein we describe their isolation, structural elucidation, possible biosynthetic pathway, and cytotoxicities.
 | ||
| Fig. 1 Chemical structures of 1–3. | ||
The EtOAc-soluble fraction of the 95% EtOH extract of A. pertyoides was subjected to repeating column chromatography (CC) on macroporous resin, silica gel, ODS, Sephadex LH-20, and semi-preparative HPLC to yield three new (1–3) and 15 known sesquiterpenoids (4–18). By comparison of the NMR and MS data with previously reported data in the literatures, 15 known compounds were characterized as macrophyllilactone F (4),10 alantolactone (5),11,12 isoalantolactone (6),11 11,13-dihydroisoalantolactone (7),13 13-hydroxy-8(αH)-eudesma-5,7(11)-dien-8,12-olide (8),14 11α,13-dihytdrosantamarin (9),6 3α-hydroxy-11αH-guaia-4(15),10(14)-dien-12,6α-olide (10),15 zaluzamin C (11),16 diaspanolide A (12),17 dihydroestafiatll (13),6 4β,14-dihydrozaluzanin C (14),18 11α,13-dihydrozaluzanin C (15),19 zauzanin C (16),20 estafiatone (17),21 and dihydroestafiatone (18).21
Pertyolide A (1) was obtained as a colorless crystal. The molecular formula was assigned as C17H24O4 by the HRESIMS ion peak at m/z 315.1579 [M + Na]+ (calcd 315.1567), implying six degrees of unsaturation.‡ The IR spectrum suggested the presence of hydroxy (3353 cm−1), γ-lactone (1766 cm−1), ketone (1705 cm−1) and olefinic (1643 cm−1) functionalities. The 1H NMR spectrum of 1 displayed diagnostic signals for two tertiary methyls [δH 0.78 (3H, s) and 2.34 (3H, s)] and two olefinic protons [δH 4.43 (1H, s) and 4.79 (1H, s)] (Table 1). The 13C NMR and DEPT spectroscopic data revealed 17 carbon resonances that were distinguished via DEPT and HSQC data to be two methyls, five methylenes (including one sp2 methylene at δC 106.5), three methines (one oxygenated at δC 77.7), and five quaternary carbons (including one oxygenated at δC 79.7, one sp2 methine at δC 149.4, one keto group at δC 210.5, and one ester carbonyl at δC 175.5) (Table 2). Its 1H and 13C NMR spectra displayed characteristic resonances of a eudesmane-type sesquiterpenoid lactone and exhibited a great similarity with those of compound 6, except for the reduction of exocyclic olefinic bond [δC 79.7; δH/δC 2.64 (1H, d, J = 17.5 Hz), 3.02 (1H, d, J = 17.5 Hz)/42.2] and the presence of an additional methyl (δH/δC 2.34 (3H, s)/32.0) and an additional ketone (δC 210.5) in 1. The HMBC correlations from H2-13 to C-11 (δC 79.7), C-12 (δC 175.5) and C-16 (210.5) and from H3-17 (δH 2.34) to C-16 (δC 210.5) linked two surplus carbons at C-13 as acetyl group. The 1H–1H COSY spectrum indicated the H-1/H-2/H-3 and H-5/H-6/H-7/H-8/H-9 proton sequences (Fig. 2). The HMBC cross-peaks between H-6/C-11, H-9/C-8, and H-9/C-7 indicated that a γ-lactone function was formed between C-8 and C-11 (Fig. 2). In the HMBC spectrum, the olefinic protons at δH 4.43 (s) and 4.79 (s) were correlated with C-4 (δC 149.4), C-3 (δC 36.9) and C-5 (δC 46.7), indicating the location of the exomethylene double bond at C-4 and proton signal assignment to H2-15. Moreover, the HMBC correlations from H3-14 to C-1, C-5, C-9, and C-10 confirmed that CH3-14 was connected to C-10. These evidences support the C17-eudesmanolide skeleton of 1 with an extended acetyl.
| No. | 1, δH (J in Hz) | 2, δH (J in Hz) | 3, δH (J in Hz) |
|---|---|---|---|
| a Data were recorded in CDCl3 at 500 MHz for 1H NMR. | |||
| 1a | 1.23, m | 1.15, m | 2.88, q (8.4) |
| 1b | 1.53, m | 1.62, m | |
| 2a | 1.26, m | 1.40, m | 2.44, m |
| 2b | 1.60, m | 1.80, m | 1.76, m |
| 3a | 2.00, m | 1.56, m | 5.56, m |
| 3b | 2.34, m | ||
| 4 | 2.46, m | ||
| 5 | 1.81, d (12.4) | 2.75, t (9.2) | |
| 6a | 1.06, q (12.4) | 4.94, d (3.5) | 4.37, t (9.2) |
| 6b | 1.42, m | ||
| 7 | 2.38, m | 3.01, dd (5.4, 3.5) | 1.94, m |
| 8a | 5.04, m | 5.13, dt (5.4, 3.0) | 1.64, m |
| 8b | 1.81, m | ||
| 9a | 1.46, dd (15.6, 4.5) | 2.14, dd (14.9, 3.0) | 2.03, m |
| 9b | 2.20, dd (15.6, 2.6) | 1.51, dd (14.9, 3.0) | 2.50, m |
| 13a | 2.64, d (17.5) | 2.65, d (17.5) | 2.62, d (16.7) |
| 13b | 3.02, d (17.5) | 2.95, d (17.5) | 2.80, d (16.7) |
| 14a | 0.78, s | 1.22, s | 4.93, s |
| 14b | 4.91, s | ||
| 15a | 4.79, s | 1.13, d (7.7) | 5.43, br s |
| 15b | 4.43, s | 5.30, br s | |
| 17 | 2.34, s | 2.33, s | 2.32, s |
| 2′ | 2.23, dd (7.1, 1.7) | ||
| 3′ | 2.12, m | ||
| 4′, 5′ | 0.97, d (6.5) | ||
| No. | 1 | 2 | 3 |
|---|---|---|---|
| a Attached protons were deduced by DEPT experiment. | |||
| 1 | 42.2a | 42.3 | 44.3 |
| 2 | 22.8 | 16.9 | 36.4 |
| 3 | 36.9 | 32.9 | 75.5 |
| 4 | 149.4 | 38.7 | 148.4 |
| 5 | 46.7 | 152.8 | 50.3 |
| 6 | 21.2 | 113.7 | 83.0 |
| 7 | 47 | 47.1 | 52.0 |
| 8 | 77.7 | 77.2 | 25.2 |
| 9 | 41.4 | 42.7 | 34.6 |
| 10 | 34.7 | 33.1 | 148.2 |
| 11 | 79.7 | 79.2 | 76.5 |
| 12 | 175.5 | 175.5 | 176.0 |
| 13 | 42.2 | 43.6 | 44.4 |
| 14 | 18.0 | 28.8 | 114.0 |
| 15 | 106.5 | 23.2 | 114.5 |
| 16 | 210.5 | 210.4 | 210.4 |
| 17 | 32.0 | 32.0 | 32.3 |
| 1′ | 173.1 | ||
| 2′ | 43.8 | ||
| 3′ | 25.9 | ||
| 4′ | 22.5 | ||
| 5′ | 22.6 | ||
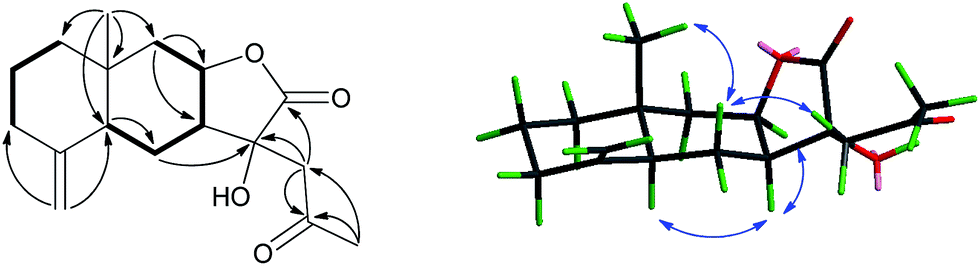 | ||
Fig. 2 Key 1H–1H COSY ( ), HMBC ( ), HMBC ( ), and NOESY ( ), and NOESY ( ) correlations of 1. ) correlations of 1. | ||
The relative configuration of 1 was deduced from the NOESY spectrum (Fig. 2) and biogenetic consideration. On the basis of biogenetic considerations, the H-7 was predicted to be α-oriented. The NOESY correlations of H-7α/H-5 and H-7α/H-8 confirmed H-5, H-7 and H-8 to be α-oriented. The NOESY correlations of H-14/H-6β and H-6β/H2-13 indicated that Me-14 and the side chain at C-11 were β-oriented, while 11-OH was α-oriented. The X-ray crystallographic data corroborated the planar structure and relative configuration of 1 as elucidated via NMR data and further allowed the assignment of its absolute configuration as 5S, 7S, 8R, 10R, 11R [Fig. 3, Flack parameter: 0.02 (7)], being a unprecedented C17 skeleton eudesmanolide.
 | ||
| Fig. 3 X-ray crystallographic structure of 1. | ||
Pertyolide B (2) was obtained as a colorless oil and exhibited the same molecular formula C17H24O4 as that of 1, as established by HRESIMS at m/z 315.1577 [M + Na]+ (calcd 315.1567), requiring six degrees of unsaturation. The IR spectrum suggested the presence of hydroxy (3440 cm−1), γ-lactone (1774 cm−1) and ketone (1718 cm−1) groups. The 1H and 13C NMR data of 2 showed a close structural resemblance to 1 (Tables 1 and 2), differing in the locations of the double bond [δH 4.94 (d, J = 3.5 Hz, H-6), δC 152.8 (C-5), 113.7 (C-6)]. The HMBC correlations from H-6 to C-4, C-7, C-8 and C-10 supported the occurrence of the double bond at C-5 and C-6 in 2 (Fig. 4), instead of the exocyclic double bond between C-4 and C-15 in 1. The H-1/H-2/H-3/H-4/H-15 and H-6/H-7/H-8/H-9 proton spin systems were deduced from the 1H–1H COSY correlations (Fig. 4). The HMBC spectrum showed the following correlations: H-6 with C-7 and C-8; H-13 with C-7, C-11, C-12 and C-16; H-17 with C-13 and C-16; H-14 to C-1, C-5, C-9 and C-10; and H-15 to C-3, C-4 and C-5. These data further supported the structure of 2.
 | ||
Fig. 4 Key 1H–1H COSY ( ), HMBC ( ), HMBC ( ), and NOESY ( ), and NOESY ( ) correlations of 2. ) correlations of 2. | ||
The relative configurations of 2 was established as shown via excellent NMR comparisons with 1 at all stereo centers, and were further validated by NOESY experiment (Fig. 4). Similar to 1, H3-14 and H2-13 were determined to be β-oriented via the NOESY correlations of H3-14/H2-13, while H-7 and H-8 were determined to be α-oriented via the NOESY correlation of H-7/H-8 and biogenetic considerations. In addition, the new chiral center of C-4 was established on the basis of the NOESY correlations of H3-15/H3-14 and H-4/H-6, and the H3-15 was assigned as β-oriented. Thus, the structure of 2 was thereby deduced and named pertyolide B.
Pertyolide C (3) gave the molecular formula C22H30O6 as determined by the HRESIMS ion at m/z 408.2387 [M + NH4]+ (calcd 408.2386) and from the 13C NMR data. The IR spectrum implied the presence of hydroxyl (3394 cm−1), ester (1729 cm−1), γ-lactone (1772 cm−1) groups. The 1H and 13C NMR spectra of 3 revealed structural fragments similar to those found in compound 11 (Tables 1 and 2). Some characteristic signals between 3 and 11 are very similar in their chemical shifts, such as the two exocyclic double bonds [δH 4.93 (1H, s, H-14a), 4.91 (1H, s, H-14b), 5.43 (1H, s, H-15a), 5.30 (1H, s, H-15b) and δC 148.2 (C-10), 114.0 (C-14), 148.4 (C-4), 114.5 (C-15)], an isovalerate moiety [δH 2.23 (2H, d, J = 7.1 Hz, H-2′), 2.12 (1H, m, H-3′), 0.97 (6H, d, J = 6.5 Hz, H-4′, 5′) and δC 173.1 (C-1′), 43.8 (C-2′), 25.9 (C-3′), 22.5 (C-4′), 22.6 (C-5′)], and one lactone ring [δH 4.37 (1H, t, J = 9.7 Hz, H-6) and δC 176.0 (C-12), 83.0 (C-6)]. However, the major changes of the chemical shifts were about C-11 and C-13. The proton and carbon signals for a methylene (δH 2.62, 2.80; δC 44.4), an oxygenated quaternary carbon (δC 76.5), and an acetyl group (δH 2.32; δC 32.3, 210.4) were observed in 3, instead of those of the Δ (ref. 11) double bond in compound 11. In addition, HMBC correlations (Fig. 5) observed between H-13 and C-11/C-16 and Me-17 and C-16/C-13, indicated the existence of a C4 side chain in 3. Moreover, an HMBC correlation between H-13 (δH 5.37) and C-12 showed that the C4 side chain was connected to C-11. In the 1H–1H COSY experiment (Fig. 5), a long fragment was deduced by the correlations of H-3/H-2/H-1 and H-5/H-6/H-7/H-8/H-9. Additionally, the HMBC correlations of H-14 to C-1 and C-9, and the correlations of H-15 to C-3 and C-5 further confirmed the planar structure of 3.
 | ||
Fig. 5 Key 1H–1H COSY ( ), HMBC ( ), HMBC ( ), and NOESY ( ), and NOESY ( ) correlations of 3. ) correlations of 3. | ||
Finally, the relative stereochemistry of 3 was established by a NOESY experiment (Fig. 5). The cis ring junction at C-1 and C-5 was confirmed from the NOESY correlation of H-1/H-5. On the other hand, H-1, H-3, H-5 and H-7 were determined to be α-oriented via the NOESY correlation of H-1/H-3, and H-5/H-7. In addition, the C4 side chain was established to be α-oriented on the basis of the key NOESY correlations between H2-13(δH 2.62, 2.80) and H-7 (δH 1.94). Thus, the structure of compound 3 was established and named pertyolide C.
Pertyolides A and B are two unprecedented C17-eudesmanolides. To date, no any similar eudesmane sesquiterpenoid has been reported with C17 skeleton. Therefore, the possible biogenetic pathway for pertyolides A and B will be an interesting topic. Our group have reported four unprecedented C17-pseudoguaianolides with an extended side chains from Inula hookeri.22 Five natural 11,16-oxetane lactones, structurally appertained to C17-guaianolides, also have been reported from Centaurea genus previously.23 The photoaddition of acetaldehyde to guaianolides could provide C17-guaianolides. Consequently, a possible biogenetic pathway for pertyolides A and B was proposed as shown in Scheme 1. In this biogenetic pathway, two precursor compounds 5 and 6 could be converted into C17 eudesmanolides 1 and 2 by an addition reaction with acetyl-CoA. To date, there is no report about that an acetyl moiety was attached to the eudesmane skeleton at C-13 by a carbon–carbon linkage. Therefore, the formation of 1–2 would provide a new insight into the structural transformation of eudesmanes.
 | ||
| Scheme 1 Plausible biogenetic pathway to pertyolides A (1) and B (2). | ||
Considering that some sesquiterpenoids from Ainsliaea species, such as ainsliatrimer A, have been reported to have significant anti-tumor activity,4,5 all the isolates were evaluated for cytotoxicity against four human tumor cell lines A549, HCT116, MGC803 and CCRF-CEM (Table 3). Only alantolactone (5) exhibited significant inhibition against A549, HCT116, MGC803 and CCRF-CEM cell lines with IC50 values of 3.56, 2.23, 2.89 and 14.67 μM, respectively. The compound 6 exhibited moderate activity toward HCT116 and MGC803 cells, with IC50 values of 14.4 and 15.1 μM, respectively. From these results, only compound 5 exhibited significant inhibitions against the tumor cell lines.
In summary, we have discovered three irregular sesquiterpene lactones named pertyolides A–C (1–3) that possess unusual C17-sesquiterpenoid frameworks and 15 known compounds from A. Pertyoides. To the best of our knowledge, 1–2 represent the first two C17-eudesmanolides with extended side chains from nature. It is noteworthy that compound 5 exhibited much stronger inhibitory activity against four tumor cell lines than any sesquiterpene lactones from A. pertyoides.
Acknowledgements
The work was supported by program NCET Foundation, NSFC (81373301, 81230090), Shanghai Leading Academic Discipline Project (B906), Key laboratory of drug research for special environments, PLA, Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (10DZ2251300), the Scientific Foundation of Shanghai China (12401900801, 13401900101), National Major Project of China (2011ZX09307-002-03) and the National Key Technology R&D Program of China (2012BAI29B06).Notes and references
- F. Bohlmamm and Z. L. Chen, Phytochemistry, 1982, 21, 2120–2122 CrossRef.
- S. Adegawa, T. Miyase and A. Ueno, Chem. Pharm. Bull., 1987, 35, 1479–1485 CrossRef CAS.
- F. Hilmi, J. Gertsch, P. Bremner, S. Valovic, M. Heinrich, O. Sticher and J. Heilmann, Bioorg. Med. Chem., 2003, 11, 3659–3663 CrossRef CAS.
- T. Dong, C. Li, X. Wang, L. Y. Dian, X. G. Zhang, L. Li, S. Chen, R. Cao, L. Li, N. Huang, S. He and X. G. Lei, Nat. Commun., 2015, 6, 6522–6533 CrossRef CAS PubMed.
- C. Li, T. Dong, Q. Li and X. G. Lei, Angew. Chem., 2014, 53, 12111–12115 CrossRef CAS PubMed.
- Z. J. Wu, X. K. Xu, H. W. Zeng, Y. H. Shen, J. M. Tian, J. Su, H. L. Li, L. Shan, L. H. Liu and W. D. Zhang, Planta Med., 2011, 77, 1545–1550 CrossRef CAS PubMed.
- Z. J. Wu, X. K. Xu, Y. H. Shen, J. Su, J. M. Tian, S. Liang, H. L. Li, R. H. Liu and W. D. Zhang, Org. Lett., 2008, 10, 2397–2400 CrossRef CAS PubMed.
- Y. Wang, Y. H. Shen, H. Z. Jin, J. J. Fu, X. J. Hu, J. J. Qin, J. H. Liu, M. Chen, S. K. Yan and W. D. Zhang, Org. Lett., 2008, 10, 5517–5520 CrossRef CAS PubMed.
- State Administration of Traditional Chinese Medicine, Medicine Herb in Chinese, Shanghai Science and Technology Press, Shanghai, 1999, vol. 7, p. 645 Search PubMed.
- B. Fu, B. N. Su, Y. Takaishi, G. Honda, M. Ito, Y. Takeda, O. K. Kodzhimatov and O. Ashurmetov, Phytochemistry, 2001, 58, 1121–1128 CrossRef CAS.
- K. T. Wang, H. T. Liu, Y. K. Zhao, X. G. Chen, Z. D. Hu, Y. C. Song and X. Ma, Talanta, 2000, 52, 1001–1005 CrossRef.
- X. G. Cheng, Q. Zeng, J. Ren, J. J. Qin, S. D. Zhang, Y. H. Shen, J. X. Zhu, F. Zhang, R. J. Chang, Y. Zhu, W. D. Zhang and H. Z. Jin, Eur. J. Med. Chem., 2011, 46, 5408–5415 CrossRef CAS PubMed.
- Y. M. Zhao, M. L. Zhang, C. H. Huo, Y. C. Gu and Q. W. Shi, Nat. Prod. Res. Dev., 2009, 21, 616–618 CAS.
- S. Z. Huang, L. B. Li, S. P. Jiang, X. L. Chen and H. J. Zhu, Helv. Chim. Acta, 2010, 93, 1808–1811 CrossRef CAS PubMed.
- M. Yang and Z. J. Jia, Chin. Chem. Lett., 2004, 15, 417–418 CAS.
- R. Mata, I. R. Cruz, B. R. Cruz, R. Bye and B. N. Timmermann, J. Nat. Prod., 2002, 65, 1030–1032 CrossRef CAS PubMed.
- S. Adegawa, T. Miyase and A. Ueno, Chem. Pharm. Bull., 1987, 35, 1479–1485 CrossRef CAS.
- B. Ferdinand and Z. L. Chen, Phytochemistry, 1982, 21, 2120–2122 CrossRef.
- X. S. Li, J. Y. Liu, J. N. Cai and P. L. Cai, Magn. Reson. Chem., 2008, 46, 1070–1073 CrossRef CAS PubMed.
- S. Z. Choi, M. C. Yang, S. U. Choi and K. R. Lee, Arch. Pharmacal Res., 2006, 29, 203–208 CrossRef CAS.
- G. N. K. Kumari, S. Masilamani, M. R. Ganesh and S. Aravind, Phytochemistry, 2003, 62, 1101–1104 CrossRef CAS.
- X. R. Cheng, J. Ren, C. H. Wang, B. Guan, J. J. Qin, S. K. Yan, H. Z. Jin and W. D. Zhang, Tetrahedron Lett., 2013, 54, 1943–1946 CrossRef CAS PubMed.
- F. A. Macías, V. M. I. Viñolo, F. R. Fronczek, G. M. Massanet and J. M. G. Molinillo, Tetrahedron, 2006, 26, 7747–7755 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The extraction scheme, spectroscopic data, bioassay methods, computational method, and crystallographic data of 1. CCDC 1405733. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra16551b |
‡ Pertyolide A (1): colorless crystal from petroleum ether/acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). m.p. 136–138 °C; [α]25D +42.0 (c 0.100, CH3OH); UV (CH3OH) λmax 202 nm; IR (KBr) νmax 3354, 2924, 2852, 1766, 1705, 1643, 1442, 1263, 1173, 1090, 887, 802, 636 cm−1; 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 315.2 ([M + Na]+), 291.0 ([M − H]−); positive HRESIMS m/z 315.1579 ([M + Na]+, calcd 315.1567). Crystal data for pertyolide A (1): C17 H24 O4: M 292.36, orthorhombic, space group P 21 21 21, a = 6.38940(10), b = 11.8924(2), c = 19.9718(4) Å, α = 90°, β = 90°, γ = 90°, V = 1517.56(5) Å3, Z = 4, μ = 0.727 mm−1, 9899 reflections measured, 2731 unique [R(int) = 0.0285]. The final wR(F2) was 0.0896 (all data). CCDC 1405733. Pertyolide B (2): colorless oil; [α]25D +1.0 (c 0.100, CH3OH); UV (CH3OH) λmax 201 nm; IR (KBr) νmax 3440, 2925, 2852, 1774, 1718, 1664, 1458, 1363, 1192, 1063, 970, 891, 667 cm−1; 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 293.2 ([M + H]+), 315.2 ([M + Na]+); positive HRESIMS m/z 315.1577 ([M + Na]+, calcd 315.1567). Pertyolide C (3): colorless oil; [α]25D +17.0 (c 0.100, CH3OH); UV (CH3OH) λmax 202 nm; IR (KBr) νmax 3394, 2924, 2852, 1772, 1729, 1647, 1466, 1367, 1292, 1259, 1169, 1074, 903 cm−1; 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 413.3 ([M + Na]+); positive HRESIMS m/z 408.2387 ([M + NH4]+, calcd 408.2386). :1). m.p. 136–138 °C; [α]25D +42.0 (c 0.100, CH3OH); UV (CH3OH) λmax 202 nm; IR (KBr) νmax 3354, 2924, 2852, 1766, 1705, 1643, 1442, 1263, 1173, 1090, 887, 802, 636 cm−1; 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 315.2 ([M + Na]+), 291.0 ([M − H]−); positive HRESIMS m/z 315.1579 ([M + Na]+, calcd 315.1567). Crystal data for pertyolide A (1): C17 H24 O4: M 292.36, orthorhombic, space group P 21 21 21, a = 6.38940(10), b = 11.8924(2), c = 19.9718(4) Å, α = 90°, β = 90°, γ = 90°, V = 1517.56(5) Å3, Z = 4, μ = 0.727 mm−1, 9899 reflections measured, 2731 unique [R(int) = 0.0285]. The final wR(F2) was 0.0896 (all data). CCDC 1405733. Pertyolide B (2): colorless oil; [α]25D +1.0 (c 0.100, CH3OH); UV (CH3OH) λmax 201 nm; IR (KBr) νmax 3440, 2925, 2852, 1774, 1718, 1664, 1458, 1363, 1192, 1063, 970, 891, 667 cm−1; 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 293.2 ([M + H]+), 315.2 ([M + Na]+); positive HRESIMS m/z 315.1577 ([M + Na]+, calcd 315.1567). Pertyolide C (3): colorless oil; [α]25D +17.0 (c 0.100, CH3OH); UV (CH3OH) λmax 202 nm; IR (KBr) νmax 3394, 2924, 2852, 1772, 1729, 1647, 1466, 1367, 1292, 1259, 1169, 1074, 903 cm−1; 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 413.3 ([M + Na]+); positive HRESIMS m/z 408.2387 ([M + NH4]+, calcd 408.2386). |
| This journal is © The Royal Society of Chemistry 2015 |