DOI:
10.1039/C5RA16532F
(Paper)
RSC Adv., 2015,
5, 98231-98240
Polyimides as metal-free catalysts for organic dye degradation in the presence peroxymonosulfate under visible light irradiation
Received
16th August 2015
, Accepted 5th November 2015
First published on 10th November 2015
Abstract
Polymer based materials, polyimides (PI) were synthesized by direct thermal condensation of melamine and pyromellitic dianhydride and characterized by scanning electron microscopy, transmission electron microscopy, X-ray diffraction, Fourier transformed infrared spectroscopy, N2 adsorption/desorption isotherms and X-ray photoelectron spectroscopy. The materials were then applied as catalysts for organic dye degradation under visible light irradiation in the presence of peroxymonosulfate (PMS). The results revealed that the metal-free catalysts can activate PMS to generate reactive radicals under visible light irradiation, leading to the efficient degradation of acid orange 7 (AO7) and other organic dyes in aqueous solution. Recycling of the catalyst indicated no obvious deactivation during the entire catalytic reaction, suggesting the good stability of metal-free polymeric photocatalysts. The intermediate products after AO7 degradation were analyzed by UV-vis spectra, ESI-MS and GC/MS. The effect of operation parameters such as the concentration of the catalyst, PMS and organic dye, and initial solution pH on the catalytic activity was also investigated. This study demonstrated a promising polymer material for organic dye degradation with PMS under visible light irradiation.
1. Introduction
Dye effluents discharged by various industries such as textile, pulp and paper, leather, food and dyestuff manufacturing, contain high organic contents, strong color, surfactants and additives. These pollutants are difficult to dispose of by traditional biological technologies and present a huge challenge to the eco-environment.1–3 In recent years, advanced oxidation technologies (AOPs) including electrochemical treatment,4 ozonation,5 photocatalysis,6 Fenton and Fenton like reagents5 have been reported as efficient alternative solutions. The processes usually produce hydroxyl radicals (HO˙) as the main reactive species with a high oxidation potential,7 and can degrade most of the biorefractory organics in dye effluents.8 Sulfate radicals (SO4−˙) based AOPs have also received much attention as a promising technique9,10 and overcomes some of the limitations of conventional AOPs. Compared to HO˙ radicals, SO4−˙ also exhibit a similar redox potential of 2.5–3.1 V and can degrade completely most organic pollutants over a wide pH range.11 Furthermore, SO4−˙ radicals have a longer half-life, mainly because of their preference for electron transfer reactions while ˙OH participates in a variety of reactions with equal preference.12 The radicals can be generated through activation of persulfate or peroxymonosulfate (PMS) by heat,13–15 UV and transition metal ions.16,17 However, UV activation and thermal activation both need added energy, and transition metal ions catalysts such as Co(II), Cu(II), Mn(II), Ni(II) and Ce(III) are toxic and difficult to recycle. In recent years, the heterogeneous activation of PMS to degrade contaminants has become a more focused area. For example, CuFe2O4 magnetic nanoparticles have been suggested as efficient heterogeneous catalysts for PMS activation.18 In our previous works, it was also found that cryptomelane-type manganese oxide octahedral molecular sieves showed excellent activity and stability for organic dyes decompositions in aqueous solution in the presence of PMS.19–21
More recently, activation of PMS to degrade contaminants by metal-free catalysts has attracted the attention of more people. For example, Huang et al. found that the addition of O3/PMS could significantly enhance the degradation of a typical organic contaminant p-CBA.22 In the study of Yang's group, granular activated carbon (GAC) was used as a catalyst to activate PMS to degrade azo dye acid orange 7 (AO7) in aqueous solution.2 Lee et al. found that carbon nanotubes (CNTs) could activate persulfates (i.e. PMS and peroxydisulfate) into reactive species that are capable of oxidizing organic compounds in water.23 Compared with metal-based catalysts, the metal-free catalysts have advantages of no heavy metal pollution, good chemical stability and environmental friendliness.
In the past few decades, polymer materials polyimides (PI) have been widely employed in the fields of aerospace, microelectronics, coatings, functional membranes, due to their excellent mechanical properties such as high mechanical strength and excellent electrical properties, high thermal stability, good chemical resistance as well as their abundant sources, low-cost fabrication and easy processibility.24–27 The crystalline PI was also used as new organic photocatalysts for H2 production from water and organic dye degradation.28 For example, metal-containing organic–inorganic composite of PI was suggested as efficient photocatalyst toward methyl orange and rhodamine B degradation.29 Sulfur-doped polyimide was reported to exhibit an enhanced photocatalytic activity for extending the absorption range of polyimid.30 However, until now there are no investigations on the photocatalytic performance of PI combining with oxidants to increases the degradation rate of organic compounds and to avoid problems caused by low oxygen concentration. In this work, we report on the advancement of PI photocatalysts for environmental application in the presence of PMS for the first time. This heterogeneous catalysis method seems to be an economically attractive and environmentally friendly oxidation technology for the treatment of organic pollutants.
2. Experimental
2.1 Preparation of PI catalyst
The PI catalyst was prepared according to the literature.28 Briefly, melamine (MA) and pyromellitic dianhydride (PMDA) with equal molar ratio (10 mmol) were mixed uniformly in an agate mortar. Then, the mixture was put into an alumina crucible with a cover, and heated to 325 °C at a heating rate of 7 °C min−1 in a muffle furnace for 4 h. The resultant solid was ground well into powder and washed. After that the sample was filtered and dried at 60 °C for 12 h to obtain the final product.
2.2 Characterization
X-ray powder diffraction (XRD) pattern was obtained on a Bruker D8 powder X-ray Diffractometer with Cu Kα radiation (λ = 0.15406 nm). The beam voltage and current used were 40 kV and 40 mA, respectively. Fourier transform infrared (FT-IR) spectra were recorded on a Bruker Vector 22 spectrometer. The sample was mixed with solid KBr, then ground into powder and dried before pressed into a wafer. The surface morphology was characterized on a Hitachi S-4800 scanning electron microscope (SEM) instrument (Hitachi Ltd., Japan) and field-emission transmission electron microscope (FETEM, Tecnai G2 F20). UV-vis absorption spectra were recorded on a Shimadzu UV-2450 spectrometer in the range of 200–800 nm.
The determination of pore size distribution, BET surface area, and pore volume was carried out with a Quanta chrome Autosorb1 at −196 °C. The samples were first degassed at 200 °C for 6 h. The surface area was obtained using the N2 adsorption isotherm with the Brunauer–Emmett–Teller equation. The average pore diameter was calculated with the Barrett–Joyner–Halenda desorption isotherm. The chemical species of the PI catalyst were investigated by X-ray photoelectron spectra (XPS) on a VG Multilab2000 spectrometer (Thermo Electron Corporation) with AlKα radiation as the exciting source (300 W). Charging effects were corrected by adjusting the binding energy of C1s to 284.6 eV.
2.3 Photocatalytic degradation experiment
Degradation experiments were performed in a 100 mL reactor at about 25 °C without irradiation or under irradiation by a 500 W xenon lamp (Beijing Trusttech Co.). A glass filter was added to allow visible light (λ > 420 nm) to pass through. After the desired amounts of AO7 and PMS in 50 mL of the aqueous solution were added into the reactor, the reaction was initialized by adding PI. Each reaction solution was constantly stirred with a PTFE coated magnetic stirrer. Since PMS is an acidic oxidant, the addition of PMS led to a significant decrease of pH, and then the experiment was conducted at acidic medium (pH 3.80, no adjustment). For studying the effect of solution pH on the rate of AO7 degradation, H2SO4 (20 mM) and NaOH (20 mM) was used to adjust the solution pH after PMS was added into the solution. For the recycling experiment, the catalyst was separated without any treatment after each recycle, and then the next reaction was started by adding a fresh solution of AO7 and PMS.
To monitor the degradation process of organic dyes, solution samples were taken out at given time intervals and measured immediately on a Varian Cary 50 Scan UV-vis spectrophotometer under the maximum absorption wavelength. For the dyes AO7, Methylene Blue (MB), Rhodamine B (RhB), Reactive Brilliant Red X-3B (X-3B) and Methyl Orange (MO), their maximum absorption wavelengths are 484 nm, 664 nm, 555 nm, 538 nm and 464 nm, respectively. For identification of degradation products, the samples were analyzed by mass spectrometry, and the experiments were performed on an Esquire LC-ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with an orthogonal geometry ESI source. Nitrogen was used as the drying (3 L min−1) and nebulizing (6 psi) gas at 300 °C. The spray shield was set to 4.0 kV and the capillary cap was set to 4.5 kV. For the analysis by GC-MS (Agilent Mass Selective Detector, GC 5973N and MS 5973N), the filtrate after reaction was dried at room temperature to obtain the solid sample, which was then dissolved in methanol and filtered before measurement. Total organic carbon (TOC) was determined by an Apollo 9000 TOC analyzer.
3. Results and discussion
3.1 Characterization of PI
X-ray diffraction patterns of the PI catalysts are shown in Fig. 1(A). The catalysts exhibit obvious characteristic peaks in the range of 10–30°, similar to that reported in literature.28 The high crystallinity of PI indicates the high regularity of polymer chains, which is likely caused by the strong π–π interactions between conjugated core units during the imidization reaction.31 The peak located at 29.5° is corresponded to the layer spacing of 0.301 nm between two-dimensional structures, which is similar to the structure of the graphene layer spacing of 0.330 nm. Fig. 1(B) displays the FT-IR spectra of PI. The observed three bands around 1770, 1720 and 724 cm−1 can be attributed to the asymmetric stretching, symmetric stretching and bending vibration absorption peak of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. The bands at 1560 and 1306 cm−1 are indicative of the character of aromatic CN stretching and breathing modes in the triazine unit. Compared with that in triazine-based frameworks of C3N4, the stretching vibration energy of the C–N bond in PI became strong because the electron-withdrawing PMDA blocks the substituent into the framework of C3N4. The sharp peak at around 805 cm−1 originates from heptazine ring units. The peak located around at 1376 cm−1 is assigned to the stretching vibration of C–N–C in the five-membered imide rings; and the other absorption bands at 1640 and 1157 cm−1 are contributed to the aromatic C–C and C–H bonds of benzene rings in dianhydride blocks.31
O, respectively. The bands at 1560 and 1306 cm−1 are indicative of the character of aromatic CN stretching and breathing modes in the triazine unit. Compared with that in triazine-based frameworks of C3N4, the stretching vibration energy of the C–N bond in PI became strong because the electron-withdrawing PMDA blocks the substituent into the framework of C3N4. The sharp peak at around 805 cm−1 originates from heptazine ring units. The peak located around at 1376 cm−1 is assigned to the stretching vibration of C–N–C in the five-membered imide rings; and the other absorption bands at 1640 and 1157 cm−1 are contributed to the aromatic C–C and C–H bonds of benzene rings in dianhydride blocks.31
 |
| | Fig. 1 (A) XRD and (B) FT-IR spectra of fresh and used PI catalysts. | |
The morphological features of PI were studied using SEM and TEM. As observed in Fig. 2(A) and (B), the as-prepared PI shows massive, aggregated morphology. In some instances, there are some bulky flakes with the size of about several hundred nanometers, in agreement with that in literature.32
 |
| | Fig. 2 (A) SEM and (B) TEM images of fresh PI catalyst. | |
To examine the surface area, as well as the pore size distribution, nitrogen adsorption/desorption analysis was conducted. The surface area was found to be 5.36 m2 g−1, which was much smaller than that of common photocatalysts or catalyst supports.33–36 The isotherms and Barrett–Joyner–Halenda pore size distributions are shown in Fig. 3(A). The isotherms show typical hysteresis, revealing the existence of mesopores, and the pore size mainly distributes in the range of 3–30 nm. The UV-vis spectrum of the PI powder is shown in Fig. 3(B). It is observed that the PI exhibits remarkable absorbance in UV region and also strong absorption of visible light within 750 nm, due to the visible-light induced transition of electrons from the valence band to the conduction band of the semiconductor. The absorption edge of PI was estimated to be 550 nm. It is found that the optical absorption edge red-shift compared with that reported in literatures,28–30,37 indicating a higher degree of polymerization and a more pronounced overlap of molecular orbitals within the material.
 |
| | Fig. 3 (A) N2 adsorption/desorption isotherm curve of PI catalyst (inset is the pore size distribution of PI) and (B) UV-vis absorption spectrum of PI catalyst. | |
The surface composition of the obtained PI was evaluated by XPS measurements. The C1s XPS spectrum in Fig. 4(A) shows two peaks centering at 284.8 eV and 287.9 eV respectively. The first peak is attributed to the surface adventitious reference carbon [38]. As for the second peak, it originates from carbon atoms bonded to three nitrogen atoms in the PI space lattice.29 The high resolution XPS spectrum of N1s shown in Fig. 4(B) can be deconvoluted into three different components. The peaks at 398.2 eV and 399.4 eV can be attributed to the sp2 hybridized aromatic nitrogen atoms involved in triazine rings (CN–C) and the structural nitrogen atoms connecting with two carbonyl (−CO) groups in the five-membered NC4 ring, respectively.30,38,39 In addition, the peak at 404.8 eV is assigned to the charging effects or positive charge localization in heterocycles.40 The O1s XPS spectrum in Fig. 4(C) exhibits a broad peak at 531.6 eV due to the CO groups, and the high binding energy peak at 537.5 eV assigned to the O–CO groups in residual PMDA monomer.41,42
 |
| | Fig. 4 High-resolution XPS spectra of C1s (A), N1s (B), O1s (C) and S2p (D) of fresh and used PI catalysts. | |
3.2 Photocatalytic performance of PI
The catalytic performance of different systems for AO7 degradation is shown in Fig. 5(A). Control experiments showed that the removal of AO7 in the PMS or PMS/Vis systems without the catalyst was negligible, indicating that PMS could not degrade the azo dye directly and not be activated by visible light irradiation. For the experiment with PI under visible light irradiation, a slight enhancement of AO7 removal was observed, in good agreement with that the catalyst can be used as photocatalyst for organic compound degradation.28–31 But the efficiency was still very low. For the PI/PMS/Vis system, a faster degradation was obtained with about 91% of the dye removed during the reaction. In comparison, when the reaction was carried out in the dark, the removal of AO7 was only 5%, implying that PI cannot activate PMS without light. The result suggested that PI could activate PMS under visible light irradiation to produce reactive species to induce strong AO7 degradation. In addition, the activity of the PI/PMS/Vis system for degradation of other organic dyes with different chemical structures such as the quinone imine dye MB, the xanthene dye RhB, the azo dye X-3B and MO was examined under similar conditions. As shown in Fig. 5(B), all the pollutants were also decomposed efficiently. These results clearly indicated that the PI/PMS/Vis is an efficient catalytic system for remediation of organic dyes wastewater.
 |
| | Fig. 5 (A) AO7 degradation with different systems and (B) degradation of other organic dyes with PI/PMS/Vis system. Conditions: PI 1.0 g L−1, PMS 0.4 g L−1, dye 20 mg L−1, 25 °C. | |
3.3 Intermediate products analysis
Representative UV-vis spectra changes observed during AO7 reaction are depicted in Fig. 6. For the solution before reaction, it shows a main absorption band at 484 nm, corresponding to the n–π* transition of the azo form, and another band at 310 nm in ultraviolet region, which is attributed to the π–π* transition of the naphthalene ring.43 Addition of PI and PMS into the aqueous solution under visible light irradiation caused the absorption bands of the dye in the visible region to decrease with time and finally to disappear, indicating the destruction of its chromophoric structure in the vicinity of the azo-linkage. At the same time, the decrease of the band in ultraviolet region was observed, due to the opening of the naphthalene ring. In addition, the absorbance at 254 nm first increased at the first 5 min and then descended gradually in the late reaction stage. The change can be attributed to the formation and degradation of naphthaquinone type intermediates.21 Although AO7 was completely destroyed within 30 min at 20 mg L−1 AO7, 1.0 g L−1 PI and 0.4 g L−1 PMS, a very low TOC removal (9.3%) was observed in the system. This was probably caused by the low dosage of PMS and the short reaction time.
 |
| | Fig. 6 Representative UV-vis spectra changes during AO7 degradation by PI/PMS/Vis system. Conditions: PI 1.0 g L−1, PMS 0.4 g L−1, AO7 20 mg L−1, 25 °C. | |
Furthermore, the intermediate products of AO7 degradation by the PI/PMS/Vis system were identified by ESI-MS and GC/MS. The results of ESI-MS(+) are shown in Fig. 7(A). At the beginning of the reaction, an intense ion of m/z 373 corresponded to AO7 [M + Na]+ was observed as expected. During oxidative treatment, the intensity of AO7 at m/z 393 decreased significantly, indicating that the dye was degraded into some intermediate products. Meanwhile, new peaks at m/z 181, 197, 213 and 253 appeared. The negative ion mode are displayed in Fig. 7(B). An intense ion of m/z 327 corresponding to AO7 [M − Na]− was observed at 0 min. After reaction the peak at m/z 173 emerged with regularly increasing of the intensity. According to the values of m/z and the suggested structures in literature,43 these products can be identified. In addition, some products were found in GC-MS spectra, as indicated in Table 1.
 |
| | Fig. 7 (A) ESI(+) and (B) ESI (−) mass spectra of AO7 solution during degradation with PI/PMS/Vis system. Conditions: PI 1 g L−1, PMS 0.4 g L−1, AO7 20 mg L−1, 25 °C. | |
Table 1 Reaction intermediates for the degradation of AO7 in the PI/PMS/Vis system identified by GC/MS
| Entry |
Retention (time/min) |
Intermediates identified by MS |
Structural formula |
| 1 |
7.482 |
Phenol |
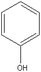 |
| 2 |
19.690 |
Phthalic acid |
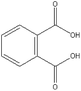 |
| 3 |
21.593 |
Coumarin |
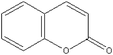 |
| 4 |
21.593 |
Benzofuran |
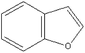 |
Based on above analysis, a schematic degradation pathway of AO7 by the PI/PMS/Vis system was proposed and is given in Scheme 1. Firstly, AO7 was decomposed to aromatic intermediates such as 1,2-naphthaquinone and p-phenolsulfonic acid through NN cleavage. Afterwards, the 1,2-naphthaquinone was further attracted by SO4−˙ or OH˙ to hydroxylated or polyhydroxylated derivatives, and finally to carboxylic acids by ring opening or the smaller molecule compounds such as benzofuran and coumarin; while p-phenolsulfonic acid could be transformed to phenol.
 |
| | Scheme 1 Proposed pathways of AO7 degradation by PI/PMS/Vis system. | |
3.4 Effect of operation parameters
To further elucidate the efficiency of PI/PMS/Vis system for AO7 degradation, the effect of AO7 concentration, PI concentration, PMS concentration, and solution pH was investigated. The influence of initial AO7 concentration at 10, 20, 30, 40 and 50 mg L−1 on the degradation rate is presented in Fig. 8. It can be seen that the degradation efficiency decreased with increasing of AO7 concentration. At 10 mg L−1, near 94% removal was achieved in about 30 min, while at 50 mg L−1, the removal was only 63% within 30 min. When increased the initial concentration of AO7, a bigger quantity of AO7 would adsorb on the surface of PI, meanwhile the active sites left for generating active species would be reduced. Furthermore, as more AO7 degraded, more intermediates generated in the reaction system. These intermediates would compete with AO7 for active species and active sites. Thus, high amount of dye in solution will require more time to achieve the same removal rate.
 |
| | Fig. 8 Influence of AO7 concentration on AO7 degradation with PI/PMS/Vis system. Conditions: PI 1.0 g L−1, PMS 0.4 g L−1, 25 °C. | |
Fig. 9 illustrates the AO7 removal with time at different PI catalyst dosages in solution. As expected, an enhancement of AO7 degradation was observed by increasing PI concentration from 0.0 to 2.0 g L−1. A 10% removal of AO7 could be reached within 30 min at 0.0 g L−1 PI loading. While complete removal could be reached at PI loading of 2.0 g L−1. The increased removal efficiency is evidently attributed to the increased availability of the active sites in the catalyst for reaction with PMS, which means a larger amount of PI could active PMS to generate more reactive radicals reacting with organic dyes in the same reaction time.
 |
| | Fig. 9 Influence of PI concentration on AO7 degradation with PI/PMS/Vis system. Conditions: PMS 0.4 g L−1, AO7 20 mg L−1, 25 °C. | |
PMS concentration has also a great impact on the treatment process efficiency and operational costs. To elucidate the role of PMS concentration on the degradation of AO7, some experiments were carried out by varying the initial PMS concentrations. As shown in Fig. 10, in the absence PMS, the AO7 removal was negligible with a removal of only 4% after 30 min. After PMS was added to the aqueous solution with a concentration of 0.02 g L−1, near 36% extent of dye was removed. The incomplete AO7 removal was probably due to lack of oxidant amount, which resulted in the deficiency of active species reacting with dye molecules. When PMS concentration increased to 0.4 g L−1, AO7 was degraded faster with a removal of 90.9% after 30 min. The increasing rate of AO7 degradation with PMS increasing can be ascribed to more active radicals produced under a higher concentration of the oxidant. Additionally, it was noticed that the further increase in PMS concentration up to 1.2 g L−1 resulted in a slight decrease of the degradation efficiency. The fact might be attributed to the decrease of the produced activate radicals due to the increase of reaction rate between the radicals and the oxidant.
 |
| | Fig. 10 Influence of PMS concentration on AO7 degradation with PI/PMS/Vis system. Conditions: PI 1.0 g L−1, AO7 20 mg L−1, 25 °C. | |
The influence of initial solution pH on AO7 degradation was studied at five different pH values from 2.54 to 8.60. As illustrated in Fig. 11, in acidic medium, a high extent of dye was removed within 30 min. At neutral condition with pH of 7.33, the degradation rate became very slow with a final removal of 38%. When the oxidation was carried out at strong alkaline medium, a much slower degradation of AO7 was observed with a final removal of 33% after 30 min. The decreased degradation efficiency might be attributed to the decreasing of adsorption ability of the catalyst for AO7, as the higher pH could decrease the electrostatic attraction between the anionic dye molecules and catalyst surfaces, which would result in lower adsorbed amount of dye and consequently in lower degradation rate. It should be also pointed out that self-dissociation of PMS mainly through non-radical pathways increases with increase in solution pH, which would additionally reduce the degradation efficiency of target contaminant.44
 |
| | Fig. 11 Influence of initial solution pH on AO7 degradation with PI/PMS/Vis system. Conditions: PI 1.0 g L−1, PMS 0.4 g L−1, AO7 20 mg L−1, 25 °C. | |
3.5 Stability of PI catalyst
Stability is one of the most important characters for a good solid catalyst in the solid–liquid reaction. To test the stability and recyclability of PI, the catalyst was collected by filtration after reaction, and the decolorization reaction was reinitiated by adding a fresh solution of AO7 and PMS. As shown in Fig. 12, the efficiency decreased during the third run in comparison with that in the first and second run, probably due to the reduction of adsorption ability. However, the catalyst was rather stable during the next runs, which could be used for five successive cycles with the removal of AO7 maintaining at about 85% after each run. The separated PI catalyst after the five reaction run was examined by XRD and FT-IR with the results shown in Fig. 1(A) and (B). In the XRD spectra, the diffraction intensity of the used PI increased, which can be ascribed to the removal of impurities during reaction. Compared with the images of the fresh catalyst, the FT-IR images of the recycled catalyst PI did not show obvious structural changes. The XPS spectra of used PI were further measured with the results shown in Fig. 4. In comparison with the fresh catalyst, the XPS spectra of C1s and N1s images of the recycled catalyst also did not show any changes. The results indicated the excellent stability of the PI catalyst in the presence of PMS with visible light irradiation.
 |
| | Fig. 12 Degradation of AO7 using the recycled PI catalyst. Condition: PI 1.0 g L−1, PMS 0.4 g L−1, AO7 20 mg L−1, 25 °C. | |
3.6 Activation mechanism of PMS on PI
PMS can be activated by UV, heat and catalysts to generate sulfate radical (SO4−˙) and hydroxyl radicals (HO˙), which possess high oxidizing ability.45 In order to understand the role of these active species in the PI/PMS/Vis system, quenching agents including ethanol (EtOH), tert-butanol (TBA), and 1,4-benzoquinone (BQ) were added into the system. The alcohols such as EtOH containing α-hydrogen can react with HO˙ and SO4−˙ radicals at significant rates, while the alcohols without an α-hydrogen such as TBA react with the two radicals very differently (fast with HO˙ but slowly with SO4−˙ radical).46 As shown in Fig. 13, in the presence of 1 mL TBA, a slight inhibition of AO7 degradation was observed. In the presence of 1 mL EtOH, the catalytic activity was obviously inhibited, and more addition of ethanol (2 mL) further decreased the rate of AO7 degradation, implying that SO4−˙ radicals were the main active species controlling the oxidation reaction, and the contribution of HO˙ radicals was little. When 0.02 mmol BQ was added into the system, an O2−˙ scavenger,47 a strong inhibiting effect was observed, suggesting the presences of O2−˙ as the active species. In addition, HSO5− might react directly with the electron hole h+ to produce SO5−˙, which can also increase the formation of photoelectron and then SO4−˙ and O2−˙ radicals. According to these analyses, the mechanism of activation of PMS by PI catalyst was proposed and is shown in Scheme 2. The adsorption and reaction of PMS in the surface of PI catalyst can be proved by the XPS spectra of S2p for PI after reaction. As indicated in Fig. 4(D), a strong peak at binding energy of 168.3 eV was observed. The peak more likely represents the signal from oxidized sulfur adsorbed by PI.48
 |
| | Fig. 13 Inhibiting effect of quenching agents on AO7 degradation with the PI/PMS/Vis system. Condition: PI 1.0 g L−1, PMS 0.4 g L−1, AO7 20 mg L−1, 25 °C. | |
 |
| | Scheme 2 The proposed mechanism of AO7 degradation in the PI/PMS/Vis system. | |
4. Conclusions
In summary, the polymer material PI was found to be an efficient and reusable photocatalyst for AO7 degradation in the presence of PMS under visible light irradiation. Nearly complete removal of AO7 and other dyes could be achieved within 30 min. The removal rate of AO7 was strongly affected by the AO7 concentration, PI amount and PMS concentration and solution pH. The catalyst can be readily recovered and reused up to five runs with almost no loss in catalytic activities. The quenching studies confirmed that sulfate radicals and hydroxyl radicals are the primary species produced during the catalytic decomposition of organic dyes. As a green oxidation process, the activation of PMS by PI under visible light irradiation may have potential application in wastewater treatment.
Acknowledgements
This work was supported by the National Science Foundation of China (21207105 and 21304072), the National Science & Technology Pillar Program (2014BAC13B02) and the Open fund from Hubei Biomass-Resource Chemistry and Environmental Biotechnology Key Laboratory (Wuhan University).
References
- M. Stoyanova, I. Slavova, S. Christoskova and V. Ivanova, Appl. Catal., A, 2014, 476, 121 CrossRef CAS.
- J. Zhang, X. Shao, C. Shi and S. Yang, Chem. Eng. J., 2013, 232, 259 CrossRef CAS.
- E. Forgacs, T. Cserháti and G. Oros, Environ. Int., 2004, 30, 953 CrossRef CAS PubMed.
- C. A. Martínez-Huitle and E. Brillas, Appl. Catal., B, 2009, 87, 105 CrossRef.
- L. Szpyrkowicz, C. Juzzolino and S. N. Kaul, Water Res., 2001, 35, 2129 CrossRef CAS PubMed.
- F. Han, V. S. R. Kambala, M. Srinivasan, D. Rajarathnam and R. Naidu, Appl. Catal., A, 2009, 359, 25 CrossRef CAS.
- J. A. Williams, W. J. Cooper, S. P. Mezyk and D. M. Bartels, Radiat. Phys. Chem., 2002, 65, 327 CrossRef CAS.
- L. Wojnárovits and E. Takács, Radiat. Phys. Chem., 2014, 96, 120 CrossRef.
- J. Criquet and N. K. V. Leitner, Chemosphere, 2009, 77, 194 CrossRef CAS PubMed.
- T. Ochiai, Y. Iizuka, K. Nakata, T. Murakami, D. A. Tryk, A. Fujishima, Y. Koide and Y. Morito, Diamond Relat. Mater., 2011, 20, 64 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705 CrossRef CAS PubMed.
- P. Shi, R. Su, S. Zhu, M. Zhu, D. Li and S. Xu, J. Hazard. Mater., 2012, 229–230, 331 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Appl. Catal., B, 2004, 54, 155 CrossRef CAS.
- S. Yang, P. Wang, X. Yang, L. Shan, W. Zhang, X. Shao and R. Niu, J. Hazard. Mater., 2010, 179, 552 CrossRef CAS PubMed.
- Y. R. Wang and W. Chu, Water Res., 2011, 45, 3883 CrossRef CAS PubMed.
- E. R. Bandala, M. A. Peláez, D. D. Dionysiou, S. Gelover, J. Garcia and D. Macías, J. Photochem. Photobiol., A, 2007, 186, 357 CrossRef CAS.
- K. H. Chan and W. Chu, Water Res., 2009, 43, 2513 CrossRef CAS PubMed.
- Y. Ding, L. Zhu, N. Wang and H. Tang, Appl. Catal., B, 2013, 129, 153 CrossRef CAS.
- S. Luo, L. Duan, B. Sun, M. Wei, X. Li and A. Xu, Appl. Catal., B, 2015, 164, 92 CrossRef CAS.
- L. Duan, B. Sun, M. Wei, S. Luo, F. Pan, A. Xu and X. Li, J. Hazard. Mater., 2015, 285, 356 CrossRef CAS PubMed.
- M. Wei, Y. Ruan, S. Luo, X. Li, A. Xu and P. Zhang, New J. Chem., 2015, 39, 6395 RSC.
- J. Cong, G. Wen, T. Huang, L. Deng and J. Ma, Chem. Eng. J., 2015, 264, 399 CrossRef CAS.
- H. Lee, H. Lee, J. Jeong, J. Lee, N. Park and C. Lee, Chem. Eng. J., 2015, 266, 28 CrossRef CAS.
- J. Cai, H. Niu, P. Zhao, Y. Ji, L. Ma, C. Wang, X. Bai and W. Wang, Dyes Pigm., 2013, 99, 1124 CrossRef CAS.
- C. Varganici, D. Rosu, C. Barbu-Mic, L. Rosu, D. Popovici, C. Hulubei and B. C. Simionescu, J. Anal. Appl. Pyrolysis, 2015, 113, 390 CrossRef CAS.
- Y. Guan, C. Wang, D. Wang, G. Dang, C. Chen, H. Zhou and X. Zhao, Polymer, 2015, 62, 1 CrossRef CAS.
- M. Hasegawa and K. Horie, Prog. Polym. Sci., 2001, 26, 259 CrossRef CAS.
- S. Chu, Y. Wang, C. Wang, J. Yang and Z. Zou, Int. J. Hydrogen Energy, 2013, 38, 10768 CrossRef CAS.
- T. Yan, M. Li, X. Wang, M. Sun, H. Liu, Q. Wei, W. Xu and B. Du, Appl. Surf. Sci., 2015, 340, 102 CrossRef CAS.
- C. Wang, Y. Guo, Y. Yang, S. Chu, C. Zhou, Y. Wang and Z. Zou, ACS Appl. Mater. Interfaces, 2014, 6, 4321 CAS.
- S. Chu, Y. Wang, Y. Guo, J. Feng, C. Wang, W. Luo, X. Fan and Z. Zou, ACS Catal., 2013, 3, 912 CrossRef CAS.
- S. Mallakpour and M. Dinari, Prog. Org. Coat., 2012, 75, 373 CrossRef CAS.
- S. Zhang, L. Zhao, M. Zeng, J. Li, J. Xu and X. Wang, Catal. Today, 2014, 224, 114 CrossRef CAS.
- X. Zhang, J. Hu and H. Jiang, Chem. Eng. J., 2014, 256, 230 CrossRef CAS.
- L. Feng, Y. Zou, C. Li, S. Gao, L. Zhou, Q. Sun, M. Fan, H. Wang, D. Wang, G. Li and X. Zou, Int. J. Hydrogen Energy, 2014, 39, 15373 CrossRef CAS.
- H. Yan, Y. Chen and S. Xu, Int. J. Hydrogen Energy, 2012, 37, 125 CrossRef CAS.
- S. Chu, C. Wang, J. Feng, Y. Wang and Z. Zou, Int. J. Hydrogen Energy, 2014, 39, 13519 CrossRef CAS.
- S. Žalenkienė, V. Krylova and J. Baltrusaitis, Appl. Surf. Sci., 2015, 325, 175 CrossRef.
- X. Yang, L. Xu, X. Yu and Y. Guo, Catal. Commun., 2008, 9, 1224 CrossRef CAS.
- M. B. Ansari, H. Jin, M. N. Parvin and S. Park, Catal. Today, 2012, 185, 211 CrossRef CAS.
- Y. Bu and Z. Chen, Electrochim. Acta, 2014, 144, 42 CrossRef CAS.
- H. J. Kim, Y. J. Park, J. Choi, H. S. Han and Y. T. Hong, J. Ind. Eng. Chem., 2009, 15, 23 CrossRef CAS.
- M. Luo, L. Lv, G. Deng, W. Yao, Y. Ruan, X. Li and A. Xu, Appl. Catal., A, 2014, 469, 198 CrossRef CAS.
- A. Rastogi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 85, 171 CrossRef CAS.
- X. He, A. A. de la Cruz and D. D. Dionysiou, J. Photochem. Photobiol., A, 2013, 251, 160 CrossRef CAS.
- W. Shi, Q. Cheng, P. Zhang, Y. Ding, H. Dong, L. Duan, X. Li and A. Xu, Catal. Commun., 2014, 56, 32 CrossRef CAS.
- Y. Tao, Q. Ni, M. Wei, D. Xia, X. Li and A. Xu, RSC Adv., 2015, 5, 44128 RSC.
- L. Zhang, D. Liu, J. Guan, X. Chen, X. Guo, F. Zhao, T. Hou and X. Mu, Mater. Res. Bull., 2014, 59, 84 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 


















