
Bis(phenolate) N-heterocyclic carbene rare earth metal complexes: synthesis, characterization and applications in the polymerization of n-hexyl isocyanate†
Abstract
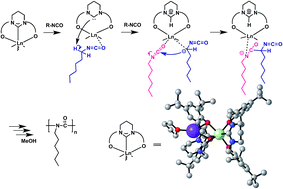
The rare earth complexes [Ln(L)2K(thf)] bearing a bis(phenolate) N-heterocyclic carbene (NHC) ligand (L = 1,3-bis[O-4,6-di-tBu-C6H2-2-CH2][C(NCH2CH2CH2N)], Ln = Nd (1), Y (2)) were synthesized in situ by the reaction of L1 with KN(SiMe3)2 and Ln[N(SiMe3)2]. The bis(phenolate) NHC precursor L1 also coordinated with rare earth metal forming bis(phenolate) pyrimidinium-bridged rare earth metal complexes [ Ln2(L1)3] (L1 = 1,3-bis[O-4,6-di-tBu-C6H2-2-CH2][CH(NCH2CH2CH2N)]+Cl−, Ln = Sm (3), Y (4)). Very high molecular weight (up to 106) and narrow molecular weight distribution (Mw/Mn = 1.7–2.3) polyhexyl isocyanate could be obtained by using the pyrimidinium based NHC rare earth metal complexes 1, 2 as well as imidazolinium based NHC rare earth complexes 5–7. The NHC complexes were found to be highly active towards the polymerization of n-hexyl isocyanate whereas non-NHC rare earth metal complexes 3, 4, 8 were inactive. The radius of rare earth metal, solvent, polymerization temperature and the structure of the ligand greatly affected the catalytic activity of polymerization. The mechanism of the initiation of the polymerization was studied and the NHC moiety played an important role in the initiation step which was evidenced via NMR analysis.
 Please wait while we load your content...
Please wait while we load your content...

