
Self-buffering and biocompatible ionic liquid based biological media for enzymatic research†
Bhupender S. Guptaa,
Mohamed Tahab and
Ming-Jer Lee*a
aDepartment of Chemical Engineering, National Taiwan University of Science and Technology, 43 Keelung Road, Section 4, Taipei 106-07, Taiwan. E-mail: mjlee@mail.ntust.edu.tw
bDepartamento de Química, CICECO, Universidade de Aveiro, 3810-193 Aveiro, Portugal
First published on 3rd December 2015
Abstract
In the present work, we have designed five new biocompatible, self-buffering ionic liquids (ILs) in which the cationic part is derived from conventional tetra-butylphosphonium (TBP) and the anionic part is derived from common biological buffers TAPS, MOPS, EPPS, CAPS, and BICINE. The new ionic liquid based biocompatible media ([TBP][TAPS], [TBP][MOPS], [TBP][EPPS], [TBP][CAPS], and [TBP][BICINE]) were found to be suitable for overcoming most of the problems associated with enzymatic research including optimum pH range, biocompatibility, and extraction. In comparison with the conventional ionic liquid based biological media, these new media do not involve an external buffering compound and maintain the optimum pH by their self-buffering capability. The buffering nature of these new ILs was confirmed by measuring their pH profiles and protonation constants in aqueous solutions at different temperatures. The biocompatibility of these new ILs was also confirmed by measuring the biological activity of the enzyme α-chymotrypsin (α-CT) in the aqueous media of these ILs. Moreover, these new ionic liquids can form an aqueous two phase system (ATPS) with a common inorganic salt such as sodium sulfate. The excellent extraction efficiency (100%) of these ionic liquid-based ATPS was observed for the extraction of enzyme α-CT, in its active form, in an IL-rich phase via a single stage extraction. Since the selected common biological buffers are biocompatible and nontoxic compounds, the ionic liquids derived from these buffer compounds could be more promising for biological research.
1. Introduction
In the past few years, the scientific world has recognized the drawbacks of the conventional harmful technologies; therefore, demands to replace these harmful processes with green and clean technologies are increasing for sustainable development. In this regard, enzymes due to their capability to turn various noxious processes into completely green and safe ones have been recognized as highly effective materials and have proved very helpful in omitting various unfriendly chemicals from conventional processes. The enzymes selectively and effectively form a transition state complex with their respective substrates, and thus not only reduce the activation energy of the reaction but also speed up the reaction to a great extent and thereby quantify the overall yield of the process. Therefore, an outstanding development has been observed in generating the application of enzyme in various industrial fields such as textile, detergent, food products, development of organic chemicals, paper, leather, natural rubber etc. In the modern time, enzymatic technology became one of the most effective and important techniques in industries and highly focused in the research and development sectors.1However the high structural complexity is the main drawback associated with the enzymes. Moreover, they remain functionally active only in their native conformation. This native or active conformation of enzymes is highly sensitive to any internal or external changes, such as temperature and pH value. In addition, the purification and separation of enzyme in the active form is also a very challenging and expensive process. Therefore, the main challenges associated with the enzymatic research are to find a suitable biocompatible biological medium that keeps the enzyme in its active form, and to design the economical and rapid extraction process.
By understanding the need of a suitable biocompatible medium, different types of biological media such as sugar and polyhydric alcohols were proposed in the past.2–6 Recently, ionic liquids, a new class of green and environmental friendly solvents, have been recognized as a suitable biocompatible medium to maintain the stability and the activity of the protein or enzyme.7–12 In comparison with the conventional molecular solvent, ionic liquids are completely composed of ions, but appear as liquid below the normal boiling point of water (100 °C).13–17 However, their degree of ionic nature depends upon the nature of the ions and respective structure of the ions.18 Due to their negligible or very low vapor pressure, they are considered as the “green” solvents and have become an alternative to the conventional volatile organic compounds (VOCs).19–23 They have many attractive and unique properties, such as high chemical and thermal stability, low flammability, high ionic conductivity, easy recycling, wide electrochemical potential window, and good solvent capability for wide range of organic or inorganic materials.24–26 Apart from these properties, their tunable nature allows us to generate specific task performing IL of our great interest.27 Due to their unique properties and tunable nature, they are highly demanded in various research and industrial fields, such as biocatalysis,28,29 polymerization,30 pharmaceutical,31 electrochemistry,32–34 gas absorption,35–38 and organic39,40 or inorganic chemistry41 etc.
Indeed, the consumption of the ionic liquids in the biological field is increasing worldwide. However, using an external buffering compound is necessary for biomolecules in conventional ionic liquid based biological media to maintain the optimum pH of the media, since the biomolecules are functionally active only in their respective pH range. Generally, aqueous ionic liquid solution, instead of the pure form of ionic liquid, is employed as a biological medium. It should be noticed that in such IL-based aqueous media, ionic liquids dissociate into their constituent ions and thus form neutral or weakly acidic or basic solutions. Hence, it is quite possible that ionic liquids may interfere with the buffering potential of the external buffering agent used in the media to maintain the optimum pH range. The problem can be more severe in the case of functional ionic liquids, such as acidic or basic catalyst. Therefore, alternative pH control methods can be highly advantageous for the biological fields involving the ionic liquid based biological media. By understanding the problems of buffering associated with the conventional ionic liquids and the challenges associated with the enzymatic research, in this study, we are introducing five new self-buffering and biocompatible ionic liquids, [TBP][TAPS], [TBP][MOPS], [TBP][EPPS], [TBP][CAPS], and [TBP][BICINE] for the biological research. These new ionic liquids were synthesized by the combination of the conventional phosphonium-ion as cation and commercial Good buffers, TAPS, MOPS, EPPS, CAPS, and BICINE, as anion. The selected Good's buffers (TAPS, MOPS, EPPS, CAPS, and BICINE) are commonly used in the various biological researches and are useful for maintaining the pH in the range of 6 to 11.42 Since these new materials are derived from Good's Buffer (GB), we call them as Good buffer ionic liquids (GBILs). These new synthesized GBILs were characterized by a series of measurement such as proton nuclear magnetic resonance (1H NMR) spectra for structure determination, Karl Fischer titration for water content determination, thermal gravimetric analysis (TGA) for thermal stability analysis, and differential scanning calorimetry (DSC) for analyzing their melting points. The buffering efficiency of the synthesized GBIL in aqueous solution was estimated by measuring their respective pH profiles as well as the experimentally determined pKa values at different temperatures. The biocompatibility of the synthesized GBIL is confirmed by measuring the biological activity of the enzyme α-chymotrypsin at pH = 8.
Even though plenty of ionic liquids for the biological research are introduced by the various research groups in past,43–50 but reports about the ionic liquids with self-buffering tendency are scares. Besides, most of the reported self-buffering ILs are not considered as a suitable candidate for the biological research due to the reactive nature of their anions. In the general practice, the phosphate based-buffer systems are highly recommended for the biological research. Probably, the high concern in the phosphate buffer system is due to its noticeable protein stabilizing tendency.51,52 But it is found that the phosphate ion forms a variety of complexes with metal ions such as magnesium (Mg), calcium (Ca) and zinc (Zn).53 Since these metal ions (Mg, Ca, and Zn) are commonly found in various proteins or enzymes, the probability of the interference of the phosphate buffer system with the respective protein is very high. Therefore, the reported new GBILs can be very useful over the conventional ILs or the buffering systems commonly used in the biological research.
Recently, the aqueous two phase systems (ATPS) for the separation and purification of the enzyme, protein, metal ions, dyes, and other biomolecule have gained great attention.54–60 Fortunately, the synthesized GBILs are found to form ATPS on introducing saturated aqueous solution of inorganic salt, sodium sulfate. Therefore, the phase boundaries data for the GBILs + salt + water ternary systems were also determined experimentally at 25 °C. The extraction efficiency (EE) for an enzyme, α-chymotrypsin (α-CT), using these GBIL-based ATPS was checked. Almost a complete extraction (100 percent) of α-CT in GBIL-rich upper phase was noticed in a single step. To further ensure that the extraction is almost 100 percent and that α-CT still remains in its active confirmation after the extraction, we measured the intrinsic fluorescence spectra and biological activity of the extracted α-CT.
The model enyzme α-chymotrypsin is well known for its structural and functional characteristics. The single polypeptide chain of α-CT is constructed by 245 amino acids and its molecular weight is 25 kDa. The structure of α-CT is composed of two juxtaposed-barrel domains with five disulfide bonds. The enzymatic site is located in the second domain of α-CT and is formed by the catalytic triad of His 57, Asp 102 and Ser 195 amino acid residues.61,62 Basically, α-CT is a serine protease enzyme, widely distributed in nature, and known to perform diverse functions.63 It is widely used to understand the folding/unfolding mechanism of protein in coslvents.64
2. Experimental section
2.1 Materials
The buffers, TAPS, MOPS, EPPS, CAPS, and BICINE, with purity greater than 0.99 in mass fraction, were purchased from Sigma Chemical Co. (USA). The enzyme α-chymotrypsin (α-CT) from bovine pancreas (type II, essentially salt free) and sodium sulfate (Na2SO4), with purity 0.99 in mass fraction, were obtained from Sigma Chemical Co. (USA). The solvents, acetonitrile with purity 0.998 in mass fraction, ethanol with purity 0.998 in mass fraction, deuterium oxide with purity 0.990 atom D, and dimethyl sulfoxide-d6 (DMSO-d6) with purity 0.999 atom D, were supplied by Sigma Aldrich Co. USA. The basic hydroxide solution of tetra-butylphosphonium ([TBP][OH], 40 wt% in H2O) was acquired from Sigma Aldrich Co. (USA). All the materials were purchased from commercial sources and are with high analytical grades; therefore, they are used without any further purification treatment. Double distilled de-ionized water used in the experiment was prepared with Nano pure-Ultra pure water purifying system at resistivity of 18.3 MΩ cm. An electronic balance (R&D, Model GR-200, Japan) with a precision of ±0.1 mg was used in the sample preparation.2.2 Synthesis and characterization of the GBILs
In the synthesis process, the aqueous solution of buffer with desired fixed number of moles was loaded in a round bottom flask, which was connected with a reflux condenser. The solution was neutralized by dropwise addition of equi-molar amount (slightly less than the buffer) of tetra-butylphosphonium hydroxide solution under constant and vigorous stirring. The reaction mixture was stirred uniformly for more than 12 hours at ambient condition. The completion of reaction mixture was ensured by checking the pH of the reaction mixture. After the completion of reaction, the mixture was then evaporated at about 50–60 °C by using a rotary evaporator (Panchum Evap., Chering Huei Co., Ltd, Taiwan) connected with a high capacity vacuum pump. To remove the excess buffer or any other impurities from the obtained product, one to one ratio mixture of ethanol and acetonitrile was added to the product and was vigorously stirred for about 1 hour at room temperature. The solution was then filtered by using microporous glass sintered crucible and the filtrate solution was dried under vacuum at the reduced pressure of 5 kPa for about 20 to 30 hours to obtained GBIL with possible low level of water content. The water content in the synthesized GBIL was measured with Karl Fischer titration. The structure of these synthesized GBILs has been defined by measuring 1H NMR spectra with a Bruker advance 500 spectrometer using tetra-methylsilane (TMS) as an internal reference. The elemental analysis for the synthesized [TBP][TAPS], as a representative GBIL, was performed with Elementar Vario EL cube (Germany) to further confirm the reported structures of these GBILs.2.3 Thermal gravimetric analysis (TGA)
The thermal profiles of the synthesized GBILs were measured by using TGA (Pyris Diamond TG-DTA) instrument. Prior to the analysis, the GBIL sample was dried in the oven to avoid the presence of moisture in the measuring sample. In each TGA measurement, the weighted sample of about 10 mg was heated from 50 °C to 600 °C at a heating rate of 10 °C min−1, under a dry nitrogen atmosphere.2.4 Differential scanning calorimetry (DSC)
The melting point of the GBIL was determined by using DSC (Perkin Elmer, Jade DSC). To avoid the moisture interference, the GBIL was dried in the oven. The dried sample of GBIL was weighted (10–15 mg) into the aluminum pans and then was heated from −50 °C to 100 °C at a heating rate of 2 °C min−1, under a dry nitrogen atmosphere.2.5 pH profile measurement
The pH profiles of the GBILs in the aqueous solutions were determined by the direct acid and base titration method. Each pH profile in the pH range of 2 to 12 was determined by using an automatic titration meter (Metrohm 888 Titrando, USA) at 25 °C. The concentrations of the stock solution of GBIL and the stock solution of titrants (sodium hydroxide or hydrochloric acid) were fixed at 0.05 M. The titration measurement was carried out in the jacketed titration cell of volume 50 cm3. During the course of experiment, the temperature of the titration cell was maintained at 25 °C by the water circulating bath (FIRSTEK, B402L, Taiwan). The temperature of the titration solution inside the cell was monitored with a precise thermometer (Model-1560, Hart Scientific Co., USA) with an uncertainty of 0.1 °C. In each experimental run, a constant volume of 30 cm3 from the stock solution of the GBIL was taken into the titration cell and was titrated against standardized acid or base under constant mixing provided by means of magnetic stirrer, in the inert nitrogen atmosphere.2.6 Protonation constant (pKa) measurement
The pKa values of each synthesized GBIL in aqueous solution at 25 °C, 30 °C and 35 °C were measured by using potentiometric method. The procedure of the measurement has been described in detail in our previous articles.65,66 The pH-value measurements at different temperatures were carried by using a jacketed and tightly closed equilibrium cell of volume 100 cm3. The temperature of the equilibrium cell was controlled at a desired value by circulating thermostatic water through the jacket of the equilibrium cell. The temperature of the titration solution inside the equilibrium cell was measured to an uncertainty of 0.1 °C by using a digital thermometer (model 1560, Hart Scientific Co., USA). In each experimental run, a titration solution 50 cm3 with GBIL concentration 0.01 mol dm−3 was titrated with standardized solution of sodium hydroxide. The ionic strength of the titration solution was fixed at 0.1 mol dm−3 by adding a calculated amount of sodium nitrate (NaNO3). The change in pH-value of the titration solution on dropwise addition of the NaOH was monitored by using an automatic titrator (Metrohm 888 Titrando, USA) equipped with a pH glass electrode. Prior to each experiment, the pH electrode was calibrated with a standard 3 point calibration method using pH 4.0, 7.0 and 10.0 standard buffer solutions. Each titration run was repeated at least three times under the inert nitrogen atmosphere in the pH range between 2.0 and 11.0. These carefully obtained potentiometric data for the investigated GBILs were further treated with the HYPERQUAD program (Version 2008)67 to calculate the pKa values of the respective GBIL. The quality of the refined value of the equilibrium constant by this program is decided on the basis of a good agreement between the calculated and experimental potentiometric data and the possible low value of the standard deviation (σ).2.7 Activity measurement
The enzymatic activity of enzyme α-CT in aqueous solution of the investigated GBILs were measured by monitoring the aqueous hydrolysis of the p-nitrophenyl acetate (PNPA). In the estimation of the catalytic activity, we used the same standard procedure as described in detail elsewhere.68,69 The progress of the PNPA hydrolysis reaction in the presence or absence of the α-CT was analyzed by monitoring the appearance of the p-nitrophenoxide (PNP−) at 400 nm, at 25 °C, and at pH = 8.0, by using UV-visible spectrophotometer (JASCO, V-550). The samples for activity measurement were prepared with 2 mg cm−3 of α-CT in 0.05 M GBILs at pH 8. Prior to the activity measurement, the prepared enzyme samples were kept about 3 to 4 hours incubation at 25 °C to attain complete equilibrium. To minimize the instantaneous aqueous hydrolysis of PNPA, the stock solution of substrate PNPA (4 mM) was prepared in acetonitrile. The value of activity was obtained from at least three repeated measurements for each sample at a given temperature. The activity is given as millimoles (mM) of PNP− formed per minute, per g of α-CT. The same procedure was used to check the activity of the extracted α-CT in GBIL-rich phase.2.8 Liquid–liquid phase boundary data
The binodal curve of GBIL-based ATPS were determined by cloud point titration method, the similar experimental procedure as suggested in the literature.70,71 The known amount of GBIL dissolved in the pre-specified amount of de-ionized water and the mixture was stirred until the solution became homogenous. Fresh saturated solution of the sodium sulfate was added drop-wisely in the aqueous GBIL solution under constant stirring until the cloud point appeared. Then de-ionize water was added drop-wisely to the cloudy solution to move into the monophasic region again. The procedure of the appearance and disappearance of cloud point was continuously repeated under the constant stirring until the cloud point was not observed on adding any amount of sodium sulfate. The weights of the GBIL and sodium sulfate in the two-phase system were measured by using an analytical balance with uncertainty of ±5 mg.2.9 Extraction of α-chymotrypsin (α-CT)
The portioning mixture containing GBIL (30 ± 1.5 wt%) + salt (13 ± 1.5 wt%) + water (57 ± 1.5 wt%) used in the extraction of α-CT was prepared with an electronic balance under ambient condition. The pH of the portioning mixture was adjusted to 8.0 by adding strong acid or base according to the requirement. At least four such portioning mixtures, each with a total weight of 5 g, were prepared in a sample tube and a fixed amount of α-CT (0.5 g kg−1) was added to each tube, in which contains extraction medium. The mixture was mixed vigorously for 15 minutes, centrifuged for 20 minutes, and then left for at least 30 minutes to ensure the complete partitioning of the enzyme α-CT into these two liquid phases. The upper and the lower phases were separated carefully and the amount of α-CT in each phase was determined by UV-visible spectrophotometer. Prior to the sample analysis, UV-visible spectrophotometer was calibrated with the standard solution of the enzyme α-CT and the obtained calibration curve was used to quantify the amount of α-CT in each co-existence phase. To avoid the interference of the salt and GBIL in protein determination, a protein-free sample containing the same amount of salt or GBIL was prepared and used as a blank. The amount of α-CT was obtained from the average of at least three repeated measurements.2.10 Fluorescence spectra measurement
After extraction, the fluorescence emission spectra for the enzyme α-CT in GBIL-rich phase and α-CT in aqueous-rich phase were measured by using a spectro-fluorophotometer (FP-8300, JASCO), with a 1.0 cm quartz-glass cell under ambient conditions. The sample of α-CT in solution containing a similar concentration of GBIL as in GBIL-rich phase was prepared and then the fluorescence spectra were measured for the comparison purpose. The spectra was obtained at an excited wavelength of 295 nm for the emission range of 300 nm to 450 nm. The excitation and emission slit width was set to 2.5 nm. Prior to the spectra measurement for the enzyme sample, the spectra for the blank solution were measured.3. Results and discussion
The new ILs were synthesized by the combination of the commonly used Good Buffer (GB) as anion and conventional phosphonium ion as cation. The selected buffers TAPS, MOPS, EPPS, CAPS, and BICINE contain acidic proton that can be easily neutralize by a suitable base. Therefore, in the synthesis process, the basic solution of phosphonium hydroxide was added drop-wisely to neutralize the aqueous solution of buffer. The reaction pathway of GBIL synthesis is shown in Scheme 1. The water content in each synthesized GBIL was measured by using Karl-Fischer titration. The results of measurements are given in Table S1 of the ESI.† The structure of each GBIL was characterized by the proton NMR spectra (1H NMR). The simplified NMR results are given in Table S1 of the ESI.† To further confirm the structure of the synthesized GBILs, we have selected [TBP][TAPS], as a representative system and conducted elemental analysis for this selected GBIL. The results of the analysis are provided in Table S1 of the ESI.† The experimentally determined percentages of the elements (carbon, hydrogen, and nitrogen) in GBIL are found very close to the analytically calculated values for the respective GBIL. The estimated structure of the GBILs are given in the Fig. 1.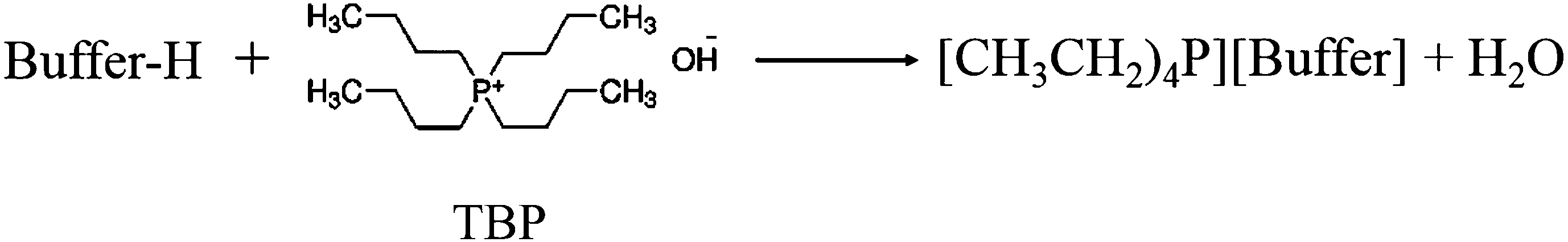 | ||
| Scheme 1 The synthesis of the GBIL. | ||
 | ||
| Fig. 1 The molecular structures of the synthesized GBILs: (a) [TBP][TAPS]; (b) [TBP][MOPS]; (c) [TBP][EPPS]; (d) [TBP][CAPS]; (e) [TBP][BICINE]. | ||
To analyze the thermal stability of the synthesized GBILs, we have measured the thermal gravimetric analysis (TGA) profile for each GBIL by using TG-DTA instrument (Pyris, Diamond). The TGA profiles of [TBP][TAPS], [TBP][MOPS], [TBP][EPPS], [TBP][CAPS], and [TBP][BIINE] are graphically presented in Fig. 2. The decomposition temperature (Td) of each GBIL was estimated from their thermal profile and listed in Table S1 of the ESI.† From the tabulated values of Td in Table S1† and the thermal profiles of GBILs in Fig. 2, it can be seen that the thermal stability of these new GBILs are in the range of 170 °C to 300 °C and highly depend on the counter anionic buffer part. The complete degradation of each GBIL was observed to occur in two successive steps. About 75% to 95% rapid mass loss was noticed just after the respective decomposition temperature in the first step. The rest of the mass loss was observed to occur slowly in the second step. The results of TGA analysis confirm that the thermal stability of these new GBILs highly depends on the selected combination of the cation and anion.
 | ||
Fig. 2 TGA thermal profiles of TBP-based GBILs: ( ), [TBP][TAPS]; ( ), [TBP][TAPS]; ( ), [TBP][MOPS]; ( ), [TBP][MOPS]; ( ), [TBP][EPPS]; ( ), [TBP][EPPS]; ( ), [TBP][CAPS]; and ( ), [TBP][CAPS]; and ( ), [TBP][BICINE]. ), [TBP][BICINE]. | ||
Furthermore, the melting point (Tm) of the GBILs was estimated by dynamic scanning calorimeter (DSC). The estimated melting point is provided in Table S1 of the ESI.† [TBP][TAPS], [TBP][MOPS], and [TBP][EPPS] exist as solid at ambient condition, while [TBP][BICINE] and [TBP][CAPS] appear as highly viscous liquids at room temperature and pressure. In general practice of biological research, the aqueous solution of ionic liquids is employed as a biological medium instead of the pure ionic liquid; therefore, those new GBILs are equally important as are the conventional less viscous ionic liquids.
To check the buffering potential of these new GBILs, we determined experimentally the pH profiles of these GBILs in aqueous solutions in the pH range of 2 to 12 at 25 °C. The pH profile curves are presented in Fig. 3. From these pH profile curves, it can be seen that each curve exhibits a region of moderate slope corresponding to the buffering region. These results confirmed that the new GBILs can be used to maintain the pH of biological media.
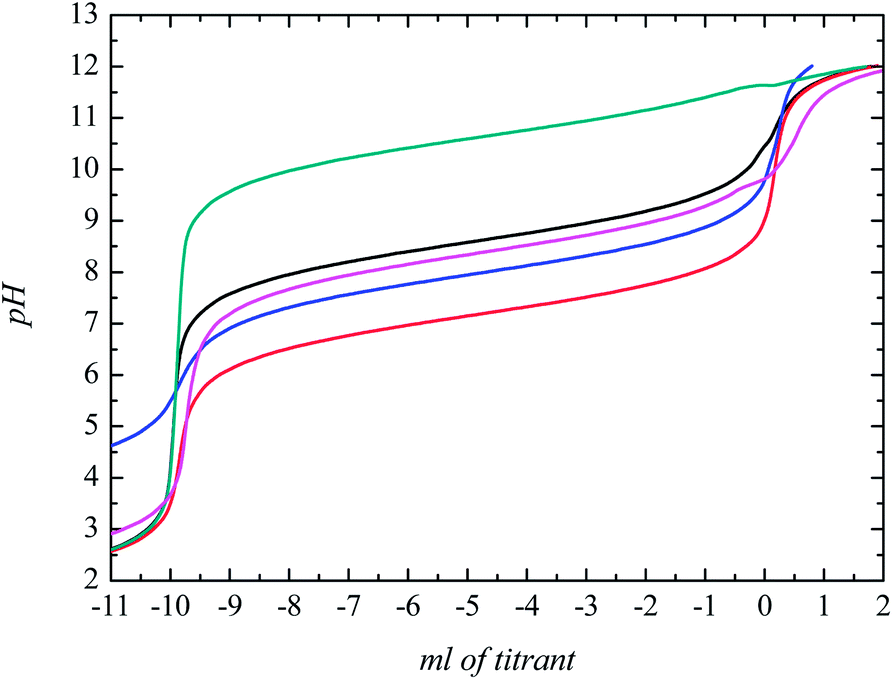 | ||
Fig. 3 pH profiles of TBP-based GBILs: ( ), [TBP][TAPS]; ( ), [TBP][TAPS]; ( ), [TBP][MOPS]; and ( ), [TBP][MOPS]; and ( ), [TBP][EPPS]; ( ), [TBP][EPPS]; ( ), [TBP][CAPS]; ( ), [TBP][CAPS]; ( ), [TBP][BICINE]. ), [TBP][BICINE]. | ||
To further define the buffering action of the synthesized GBILs, we have measured the pKa (protonation constant) of each GBIL at various temperatures (25 °C, 30 °C and 35 °C) and at ionic strength of 0.1 mol dm−3 maintained by NaNO3. On the basis of the pH-metric data, the pKa value of each GBIL was estimated with the HYPERQUAD program (version 2008).67 The calculated results are compiled in Table 1. The titration curve for [TBP][TAPS] at 25 °C, 30 °C and 35 °C along with the best fitting from HYPERQUAD is presented in Fig. 4 as a representative system. We obtained the similar titration curves for the other investigated GBILs; however, for the sake of simplicity, we do not present all the titration curves. As seen from the comparison of the best fitting values from HYPERQUAD with the experimental data in Fig. 4 and the tabulated values of the standard deviation in Table 1, the agreement is excellent. The selected Good's buffers, TAPS, MOPS, EPPS, CAPS, and BICINE, acquire two protonation constants. The first protonation constant (pKa1) represents the dissociation of sulfonic group in TAPS, MOPS, and CAPS, and the dissociation of the carboxylic group in BICINE, respectively. The second protonation constant (pKa2) is associated with the dissociation of the amine group. The second protonation constant (pKa2) is noticed to be responsible for buffering action of buffer. The pKa2 values of the corresponding Good's buffer, TAPS, MOPS, EPPS, CAPS, and BICINE in aqueous solution at 25 °C and at identical to our experimental conditions are taken from the literature42,66 and are compiled in Table 1, for the comparison purpose. The order of pKa values of these GBILs is as follows CAPS > TAPS > BICINE > EPPS > MOPS. This trend of protonation constant for the GBILs is identical with the pKa2 values of the corresponding Good's buffer. Furthermore, the pKa values of the investigated GBILs decrease with the increase of temperature from 25 °C to 35 °C. The observed trend possibly due to the enhancement of the dissociation with the rise in temperature. These pKa values of the GBILs further support our expectation that the GBILs in addition to provide suitable biocompatible media for the enzymatic or biological research, also suitable to maintain the required pH of the media in the range of 6 to 11 by their self-buffering nature.
| GB | pKa2a (25 °C) | GBILs | pKa (25 °C) | σ | pH range | pKa (30 °C) | σ | pH range | pKa (35 °C) | σ | pH range |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a pKa2 values of the Good's Buffer (GB) at 25 °C and at I = 0.1 mol dm−3.b Ref. 66.c Ref. 42. | |||||||||||
| TAPS | 8.33b | [TBP][TAPS] | 8.380 | 0.016 | 2.44–11.30 | 8.146 | 0.043 | 2.38–10.81 | 8.077 | 0.014 | 2.42–9.44 |
| MOPS | 7.17b | [TBP][MOPS] | 6.972 | 0.015 | 2.41–11.01 | 6.894 | 0.053 | 2.31–10.85 | 6.853 | 0.021 | 2.38–9.21 |
| EPPS | 7.87c | [TBP][EPPS] | 7.792 | 0.017 | 2.41–11.01 | 7.722 | 0.060 | 2.21–10.93 | 7.680 | 0.063 | 2.12–10.66 |
| CAPS | 10.39c | [TBP][CAPS] | 10.325 | 0.024 | 2.41–11.44 | 9.935 | 0.058 | 2.31–11.00 | 9.776 | 0.033 | 2.38–10.53 |
| BICINE | 8.22c | [TBP][BICINE] | 8.154 | 0.012 | 2.43–11.12 | 8.040 | 0.044 | 2.34–10.69 | 7.953 | 0.050 | 2.38–10.46 |
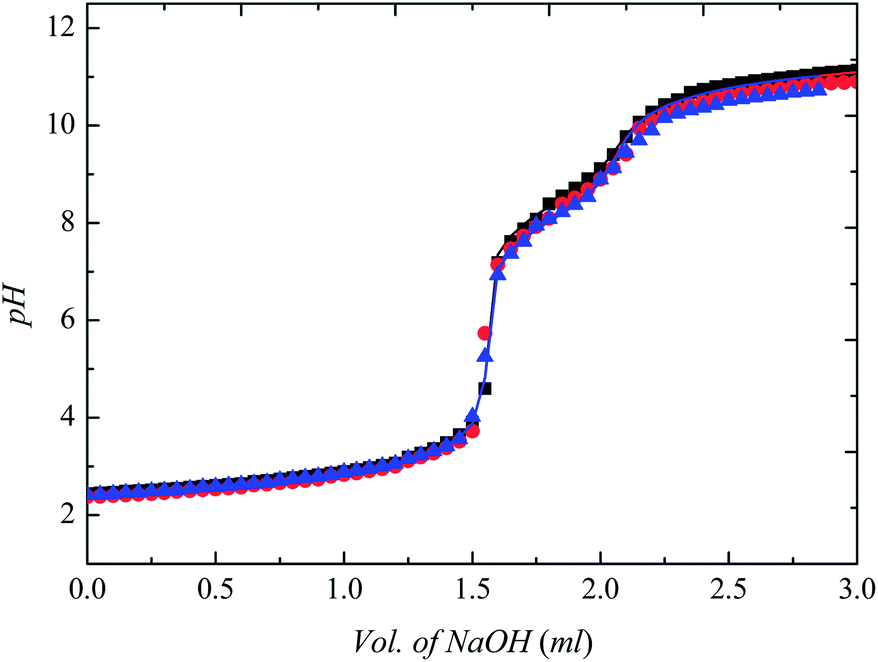 | ||
Fig. 4 The representative titration curves for [TBP][TAPS] at 25 °C, 30 °C, and 35 °C with 0.1 mol dm−3 of NaNO3: ( ), at 25 °C, ( ), at 25 °C, ( ), at 30 °C and ( ), at 30 °C and ( ), at 35 °C. The smooth line represents the corresponding best fitting from the HYPERQUAD 2008. ), at 35 °C. The smooth line represents the corresponding best fitting from the HYPERQUAD 2008. | ||
To confirm GBILs candidature as a suitable biocompatible medium for the enzymatic or biological research, we have analyzed the catalytic activity of α-CT, as a model enzyme, in the aqueous solution of the synthesized GBILs. Generally, the selected Good buffers (TAPS, MOPS, EPPS, CAPS, and BICINE) are expected to be biocompatible, nontoxic, and green compounds. This expectation is in agreement with our previous reports,72–74 in which we have observed that the buffers, TAPS, MOPS, and EPPS, provide a biocompatible medium to the protein BSA and protect the native structure of BSA against thermal denaturation in 0.5 M and 1.0 M aqueous buffer solutions. Recently, new GB-based ILs have been developed by pairing five Good's buffer anions (GBs = tricine, TES, MES, HEPES, and CHES) and 1-ethyl-3-methylimidazolium, tetramethylammonium, tetraethylammonium, tetrabutylammonium and cholinium cations,43–45 and their toxicities have been evaluated. The results of these reports have revealed that GB-ILs, in general, are even less toxic than their counterparts.43–45 It is thus expected that the addition of phosphonium ion to the selected Good's buffers may not change their inherent biocompatible nature.
However, to further confirm our expectation, we have measured the enzymatic activity of α-CT in aqueous solutions of synthesized GBILs. The enzyme is catalytically active only in its native conformation. If the GBILs will have any negative or non-biocompatible effect on the active conformation of the enzyme, it will lose its enzymatic activity to catalyze the reaction. α-CT is found to be suitable for catalyzing the hydrolysis of p-nitrophenylacetate (PNPA).75,76 Therefore, in order to check the biocompatibility of the GBILs-based media for α-CT, we have analyzed the progress of α-CT catalyzed hydrolysis of PNPA at 25 °C and at pH = 8. For the comparison purpose, the activities of α-CT were also measured in aqueous solution of MOPS buffer at the same experimental condition as for the GBILs. The results of the activity measurements are given in Table 2. As can be seen from Table 2, the presence of the GBIL makes α-CT more active in comparison with the MOPS buffer. The experimental results clearly show that these new GBILs are biocompatible in nature and assist in the active conformation of the enzyme more effectively in comparison with the conventional buffer. Activity enhancement and biocompatibility of α-CT in some conventional ionic liquids is also reported in the literature. However, the examination in the conventional ILs always required an external buffer compound in the media to maintain the optimum pH. In the conventional IL-based enzymatic media, the functional IL is used in a very high concentration, while the buffering compound is used in comparatively very low concentration. Therefore, it is more likely that cation or anion of IL interferes with the buffering action of the external buffer. Since in the present case, the inherent buffering nature of the GBILs is used to maintain the required pH of the media; therefore, the GBIL-based buffering system can be proved more effective. Assuredly GBILs not only are biocompatible but also avoid using an external buffering compound and provide effective pH control to the biological or enzymatic media. Therefore, in comparison with the traditional biocompatible ionic liquids, these new GBILs can be more promising for the biological or enzymatic research. The activities of α-CT in GBIL solutions follow the order of [TBP][TAPS] > [TBP][EPPS] > [TBP][MOPS] > [TBP][BICINE].
| Compound | Activity |
|---|---|
| MOPS | 5.689 |
| [TBP][TAPS] | 9.582 |
| [TBP][MOPS] | 8.964 |
| [TBP][EPPS] | 9.572 |
| [TBP][BICINE] | 6.982 |
In addition to the suitable biocompatible medium and the buffering agent, we have found that all the investigated tetra-butylphosphonium-based ionic liquids, [TBP][TAPS], [TBP][MOPS], [TBP][EPPS], [TBP][CAPS], and [TBP][BICINE] have tendency to form aqueous two-phase system (ATPS) by adding saturated solution of sodium sulfate. In such ATPS, the upper phase is found rich in GBIL and the lower phase is found enriched in salt. Since many enzymes are recognized highly sensitive toward internal or external stress, unstable, and thus lose their enzymatic activity during the separation and purification process. Thus, the development of the new and biocompatible separation and purification methods for enzymes or proteins are gaining great importance. In this aspect, the ATPS-based extraction process is found very advantageous. Such extraction process occurs not only under a mild condition but also are very rapid, which prevent the denaturation and inactivation of enzymes or proteins. A number of research articles77–80 focus on the economical and rapid extraction of valuable materials such as drug, metal ions, amino acids, proteins, alkaloid, and enzymes by using IL-based ATPS. By considering the importance and requirement of the ATPS in the extraction and purification processes, we have determined experimentally the binodal curve (phase boundaries) data for these newly found GBIL-based ATPS under ambient conditions. The binodal curve data was measured by the well-known cloud point titration method. The experimentally obtained phase boundaries data for each GBIL-based ATPS are tabulated in Table S2 of the ESI† and are graphically presented in Fig. 5. It can be seen from Fig. 5, each ATPS system is unique in the behavior and has separate phase boundaries. The upper region, just next to the boundary of the phase diagram, represents the two phase regions. After measuring the liquid–liquid phase boundaries for each of these GBIL-based ATPS, we are interested in analyzing the extraction efficiency of these ATPS for enzyme. For this purpose, we also selected α-CT as a model enzyme and checked the extraction efficiency via the GBIL-based ATPS under the ambient condition. We have chosen α-CT as a model enzyme because of its well-known industrial importance and unique functional characteristics. The ATPS used in the extraction of α-CT at pH 8.0 was formulated with GBIL (30 ± 1.5 wt%) + salt (13 ± 1.5 wt%) + water (57 ± 1.5 wt%). The required pH of the GBIL-based ATPS systems is maintained by the self-buffering potential of the GBILs, instead of by using external buffer compounds. We did not check the extraction efficiency of the [TBP][CAPS]-based ATPS system because it is unable to maintain the required experimental pH value (8.0). The extraction efficiency (EE) of investigated GBIL-based ATPS for α-CT is calculated by the following equation:

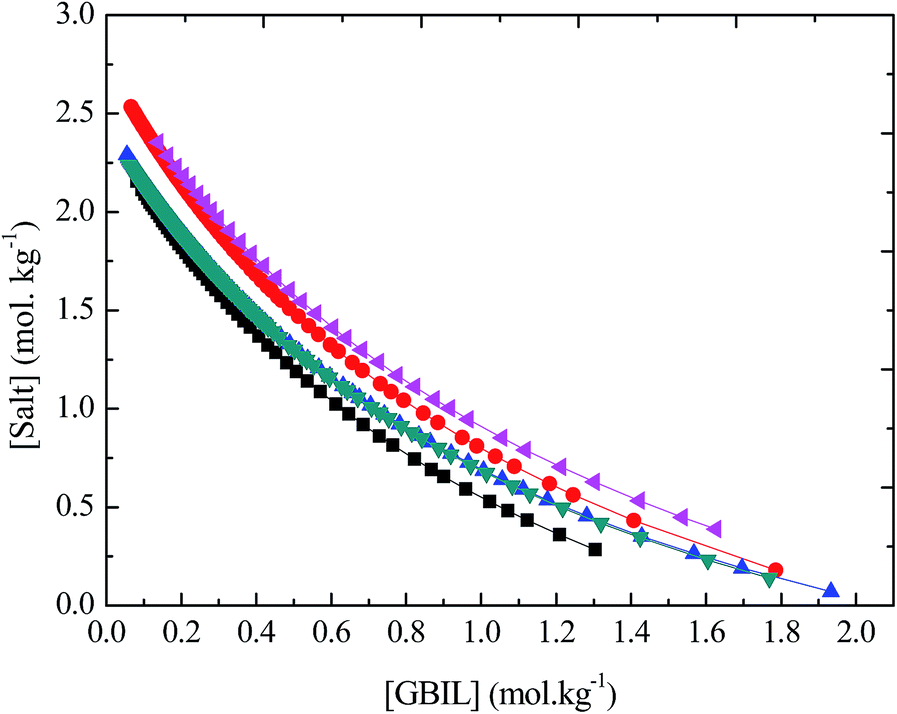 | ||
Fig. 5 Liquid–liquid phase boundaries of the systems composed of GBIL + Na2SO4 + H2O at 25 °C and atmospheric pressure: ( ), [TBP][TAPS]; ( ), [TBP][TAPS]; ( ), [TBP][MOPS]; ( ), [TBP][MOPS]; ( ), [TBP][EPPS]; ( ), [TBP][EPPS]; ( ), [TBA][CAPS]; ( ), [TBA][CAPS]; ( ), [TBA][BICINE]. ), [TBA][BICINE]. | ||
| GBIL | wIL (%) | wsalt (%) | wwater (%) | EE (%) | Activity |
|---|---|---|---|---|---|
| [TBP][TAPS] | 28.57 | 14.50 | 56.93 | 100 | 35.401 |
| [TBP][MOPS] | 29.41 | 14.85 | 55.74 | 100 | 17.366 |
| [TBP][EPPS] | 29.01 | 14.59 | 56.40 | 100 | 14.847 |
| [TBP][BICINE] | 28.08 | 14.15 | 57.77 | 97.5 | 72.081 |
To further analyze the structure of the α-CT in GBIL-rich phase and to ensure the complete extraction of α-CT in GBIL-rich phase, we have used the fluorescence method. Since any changes in the surrounding or molecular environment of the fluorophore groups can be directly studied from their fluorescence behavior.81 Therefore, the florescence technique is highly recommended for the identification of the structural configuration of enzymes or proteins in solution.82,83 The proteins or enzymes with fluorophore groups such as aromatic amino acid residues like tryptophan (Trp), phenylalanine (Phe), and tyrosine (Tyr) are noticed highly sensitive to the fluorescence spectroscopy. Specifically, it is found that on the exiting protein at the wavelength of 295 nm, the contribution from Phe or Tyr in the fluorescence spectra of respective protein became the minimum and only Trp residue contributes to the spectra. The enzyme α-CT is known to have eight tryptophan residues such as Trp27, Trp29, Trp51, Trp141, Trp172, Trp207, Trp215, and Trp237. Therefore, to understand the exact conformation of enzyme before and after the extraction with GBIL, we have measured the fluorescence spectra for α-CT in 30 percent GBIL and for α-CT in GBIL-rich phase after the extraction at pH = 8.0 and at the excitation wavelength of 295 nm. To ensure the complete extraction of α-CT, the fluorescence spectra of α-CT in GBIL-rich phase are compared with the spectra of α-CT in the salt-rich phase after extraction. The results of measurement for α-CT in [MOPS][TBP] is given in Fig. 6 and for α-CT in rest of the investigated GBILs are presented in Fig. S1(a–c) of the ESI.† Generally, the shifting in the fluorescence peaks either towards a higher wavelength or a lower wavelength is considered due to the change in the structure of protein. The change in the fluorescence intensity of the peak is considered due to the change in the fluorophore surrounding caused by the solvent. From these Fig. 6 and (S1(a–c)†), it can be seen that in comparison with the peak of α-CT in 30% GBIL (before extraction), the peak of α-CT in the GBIL-rich phase after the extraction remain intact without finding blue or red shift. However, a minor change in the fluorescence intensity of α-CT after extraction was noticed. Probably, the change in intensity is caused by the high polarity of the GBIL-rich phase. Since the fluorescence spectra of α-CT in the GBIL-rich phase after the extraction are found almost identical to the spectra of α-CT in GBIL before the extraction, it is suggested that after the extraction, enzyme remain in its active conformation. In addition, no fluorescence peak of α-CT was observed for the sample taken from the lower salt-rich phase. These findings further confirm the complete extraction of the enzyme from aqueous solutions to the upper phase via GBIL-based ATPS.
 | ||
| Fig. 6 Fluorescence spectra of the α-CT in [TBP][MOPS]-rich phase (-●-), salt-rich phase (-▲-) and in 30% [TBP][MOPS] (-■-). | ||
To support our findings from the fluorescence and be more certain that enzyme remain in the active conformation after the extraction, we have measured their catalytic activity to hydrolyze the p-nitrophenyl acetate (PNPA) at 25 °C and pH 8.0. The results of measurement are presented in Table 3. These results strongly confirmed that enzyme α-CT in GBIL-rich region still remains in the active conformation. From this series of study, it is concluded that these new GBIL-based ATPS can achieved complete extraction of active α-CT in a single step. Moreover, the enhanced catalytic activity of the α-CT in GBIL-rich phase manifests the tendency of the GBILs to support the active confirmation of the enzyme.
4. Conclusion
In this study, we have synthesized five new self-buffering and biocompatible ionic liquids for enzymatic research. These ionic liquids are derived from commercially available materials. The buffering nature of these new GBILs is ensured by measuring their pH profiles in aqueous solution. To further characterize their buffering nature experimental values of their protonation constants in aqueous solution were determined at 25 °C, 30 °C, and 35 °C. The results of measurement indicate that each new GBIL can be used as a buffer to maintain the optimum pH value in enzymatic reactions. Thus, these GBILs can be more useful in comparison with the conventional biocompatible ionic liquids because they are not only suitable for providing a similar biocompatible medium for the biological or enzymatic research but also avoid using external buffering compounds in the medium. Furthermore, the complete extraction of α-CT in its active form through the GBIL-based ATPS has been proved their promising nature for the enzymatic or biological research.Acknowledgements
The authors gratefully acknowledge the financial support from Ministry of Science & Technology, Taiwan, through NSC103-2811-E011-005 and NSC 102-2221-E011-137-MY3.Notes and references
- O. Kirk, T. V. Borchert and C. C. Fuglsang, Curr. Opin. Biotechnol., 2002, 13, 345–351 CrossRef CAS PubMed.
- R. P. Frigon and J. C. Lee, Arch. Biochem. Biophys., 1972, 153, 587–589 CrossRef CAS PubMed.
- T. Arakawa and S. N. Timasheff, Biochemistry, 1982, 21, 6536–6544 CrossRef CAS PubMed.
- A. H. Doux, J. F. Willart, L. Paccou, Y. Guinet, F. Affouard and A. Lerbret, et al., J. Phys. Chem. B, 2009, 113, 6119–6126 CrossRef PubMed.
- C. Tanford, C. E. Buckley, P. K. De and E. P. Lively, J. Biol. Chem., 1962, 237, 1168–1171 CAS.
- S. Y. Gerlsma, J. Biol. Chem., 1968, 243, 957–961 CAS.
- T. Vasantha, P. Attri, P. Venkatesu and R. S. R. Devi, J. Phys. Chem. B, 2012, 116, 11968–11978 CrossRef CAS PubMed.
- D. H. Dagade, K. R. Madkar, S. P. Shinde and S. S. Barge, J. Phys. Chem. B, 2013, 117, 1031–1043 CrossRef CAS PubMed.
- M. Bekhouche, L. J. Blum and B. Doumeche, J. Phys. Chem. B, 2012, 116, 413–423 CrossRef CAS PubMed.
- K. Rawat and H. B. Bohidar, J. Phys. Chem. B, 2012, 116, 11065–11074 CrossRef CAS PubMed.
- H. Yan, J. Wu, G. Dai, A. Zhong, H. Chen and J. Yang, et al., J. Lumin., 2012, 132, 622–628 CrossRef CAS.
- Y. Akdogan, M. J. N. Junk and D. Hinderberger, Biomacromolecules, 2011, 12, 1072–1079 CrossRef CAS PubMed.
- K. R. Seddon, A. Stark and M. J. Toress, Pure Appl. Chem., 2000, 72, 2275–2287 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- R. Sheldon, Chem. Commun., 2001, 23, 2399–2407 RSC.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS.
- D. R. MacFarlane, M. Forsyth, E. I. Izgorodina, A. P. Abbott, G. Annata and K. Fraser, Phys. Chem. Chem. Phys., 2009, 11, 4962–4967 RSC.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398–2399 CrossRef CAS PubMed.
- J. D. Holbrey and R. D. Rogers, ACS Symposium Series 818, American Chemical Society, Washington, DC, 2002 Search PubMed.
- S. A. Forsyth, D. R. MacFarlane, R. J. Thomson and M. V. Itzstein, Chem. Commun., 2002, 7, 714–715 RSC.
- C. J. Adams, ACS Symposium Series 818, American Chemical Society, Washington, DC, 2002 Search PubMed.
- A. J. Carmichael, M. Deetlefs, M. J. Earle, U. Frohlich and K. R. Seddon, ACS Symposium Series 856, American Chemical Society, Washington, DC, 2003 Search PubMed.
- Y. Chauvin, L. Mussmann and H. Olivier, Angew. Chem., Int. Ed. Engl., 1995, 34, 2698–2700 CrossRef CAS.
- Y. Chauvin, Actual. Chim., 1996, 44–46, 0151–9093 Search PubMed.
- L. Magna, G. P. Niccolai, Y. Chauvin and J. M. Basset, Organometallics, 2003, 22, 4418–4425 CrossRef CAS.
- A. García, L. C. Torres-González, K. P. Padmasree, M. G. Benavides-Garcia and E. M. Sánchez, J. Mol. Liq., 2013, 178, 57–62 CrossRef.
- S. Park and R. J. Kazlauskas, Curr. Opin. Biotechnol., 2003, 14, 432–437 CrossRef CAS PubMed.
- F. V. Rantwijk, R. M. Lau and R. A. Sheldon, Trends Biotechnol., 2003, 21, 131–138 CrossRef PubMed.
- P. Kubisa, Prog. Polym. Sci., 2004, 29, 3–12 CrossRef CAS.
- I. M. Marrucho, L. C. Branco and L. P. N. Rebelo, Annu. Rev. Chem. Biomol. Eng., 2014, 5, 527–546 CrossRef CAS PubMed.
- W. W. Tu, J. P. Lei and H. X. Ju, Chem.–Eur. J., 2009, 15, 779–784 CrossRef CAS.
- R. D. Rogers and K. R. Seddon, ACS Symposium Series 818, American Chemical Society: Washington, DC, 2002 Search PubMed.
- H. Xu, H. Y. Xiong, Q. X. Zeng, L. Jia, Y. Wang and S. F. Wang, Electrochem. Commun., 2009, 11, 286–289 CrossRef CAS.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- J. L. Anthony, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2002, 106, 7315–7320 CrossRef CAS.
- R. E. Baltus, R. M. Counce, B. H. Culbertson, H. Luo, D. W. DePaoli, S. Dai and D. C. Duckworth, Sep. Sci. Technol., 2005, 40, 525–541 CrossRef CAS.
- C. M. Wang, G. K. Cui, X. Y. Luo, Y. J. Xu, H. R. Li and S. Dai, J. Am. Chem. Soc., 2011, 133, 11916–11919 Search PubMed.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS.
- S. Ikegami and H. Hamamoto, Chem. Rev., 2009, 109, 583–593 CrossRef CAS PubMed.
- M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed. Engl., 2004, 43, 4988–4992 CrossRef CAS PubMed.
- R. N. Roy, L. N. Roy, J. G. Grant, M. P. Cummins, B. J. Tabor III, S. J. Richard, C. A. Himes, B. R. Lively, P. L. Blackwell and A. N. Simon, J. Solution Chem., 2002, 31, 861–873 Search PubMed.
- M. Taha, F. A. e. Silva, M. V. Quental, S. P. M. Ventura, M. G. Freire and J. A. P. Coutinho, Green Chem., 2014, 16, 3149–3159 RSC.
- M. Taha, M. R. Almeida, F. A. e. Silva, P. Domingues, S. P. M. Ventura, J. A. P. Coutinho and M. G. Freire, Chem.–Eur. J., 2015, 21, 4781–4788 CrossRef CAS PubMed.
- M. Taha, M. V. Quental, I. Correia, M. G. Freire and J. A. P. Coutinho, Process Biochem., 2015, 50, 1158–1166 CrossRef CAS.
- D. R. MacFarlane, R. Vijayaraghavan, H. N. Ha, A. Izgorodin, K. D. Weaver and G. D. Elliott, Chem. Commun., 2010, 46, 7703–7705 RSC.
- G. Ou, J. Yang, B. He and Y. Yuan, J. Mol. Catal. B: Enzym., 2011, 68, 66–70 CrossRef CAS.
- L. Xu, G. Ou and Y. Yuan, J. Organomet. Chem., 2008, 693, 3000–3006 CrossRef CAS.
- S. C. Matias, A. Rocha, R. Teixeira, L. J. P. Fonseca and N. M. T. Lourenço, RSC Adv., 2014, 4, 15597–15601 RSC.
- G. N. Ou, M. X. Zhu, J. R. She and Y. Z. Yuan, Chem. Commun., 2006, 46, 26–4628 Search PubMed.
- M. G. Freire, A. F. M. Claudio, J. M. M. Araujo, J. A. P. Coutinho, I. M. Marrucho, J. N. C. Lopes and L. P. N. Rebelo, Chem. Soc. Rev., 2012, 41, 4966–4995 Search PubMed.
- N. E. Good, G. D. Winget, W. Winter, T. N. Connolly, S. Izawa and R. M. M. Singh, Biochemistry, 1966, 5, 467–477 CrossRef CAS PubMed.
- P. A. Albertsson, Nature, 1956, 177, 771–774 CrossRef CAS PubMed.
- T. Agasoster, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 716, 293–298 CrossRef CAS.
- M. Rito-Palomares, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 807, 3–11 CrossRef CAS PubMed.
- C. Branden and J. Tooze, Introduction to Protein Structure, Garland Publishing, Taylor & Francis, New York, 1999 Search PubMed.
- N. J. Bridges, K. E. Gutowski and R. D. Rogers, Green Chem., 2007, 9, 177–183 RSC.
- R. D. Rogers, A. H. Bond, C. B. Bauer, J. Zhang, S. D. Rein, R. R. Chomko and D. M. Roden, Solvent Extr. Ion Exch., 1995, 14, 698–713 Search PubMed.
- P. Madeira, C. A. Reis, A. E. Rodrigues, L. Mikheeva, A. Chait and B. Zaslavsky, J. Chromatogr. A, 2011, 1218, 1379–1384 CrossRef CAS PubMed.
- M. Dilip, P. Venkateswaran and K. Palanivelu, J. Environ. Sci. Eng., 2005, 47, 176–181 CAS.
- T. E. Creighton, Proteins, Structures and Molecular Properties, ed. W. H. Freeman, New York, 1993 Search PubMed.
- C. Branden and J. Tooze, Introduction to Protein Structure, Garland Publishing, Taylor & Francis, New York, 1999 Search PubMed.
- P. Attri, P. Venkatesu and A. Kumar, Phys. Chem. Chem. Phys., 2011, 13, 2788–2796 RSC.
- V. Y. Levitsky, A. A. Panova and V. V. Mozhaev, Eur. J. Biochem., 1994, 219, 231–236 CrossRef CAS PubMed.
- M. Taha, B. S. Gupta and M. J. Lee, J. Chem. Eng. Data, 2011, 56, 3541–3551 CrossRef CAS.
- B. S. Gupta, M. Taha and M. J. Lee, J. Solution Chem., 2013, 42, 2296–2309 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- P. Attri, P. Venkatesu and M. J. Lee, J. Phys. Chem. B, 2010, 114, 1471–1478 CrossRef CAS PubMed.
- B. S. Gupta, M. Taha and M. J. Lee, RSC Adv., 2014, 4, 51111–51116 RSC.
- M. G. Freire, C. M. S. S. Neves, J. N. Canongia Lopes, I. M. Marrucho, J. A. P. Coutinho and L. P. N. Rebelo, J. Phys. Chem. B, 2012, 116, 7660–7668 CrossRef CAS PubMed.
- T. Mourão, A. F. M. Cláudio, I. Boal-Palheiros, M. G. Freire and J. A. P. Coutinho, J. Chem. Thermodyn., 2012, 54, 398–405 CrossRef.
- B. S. Gupta, M. Taha and M. J. Lee, Process Biochem., 2013, 48, 1686–1696 CrossRef CAS.
- M. Taha, B. S. Gupta, I. Khoiroh and M. J. Lee, Macromolecules, 2011, 44, 8575–8589 CrossRef CAS.
- B. S. Gupta, M. Taha and M. J. Lee, Phys. Chem. Chem. Phys., 2015, 17, 1114–1133 RSC.
- N. Spetri, F. Alfani, M. Cantarella, F. D. Amico, R. Germani and G. Savelli, J. Mol. Catal. B: Enzym., 1999, 6, 99–110 CrossRef.
- P. Viparelli, F. Alfani and M. Cantarella, J. Mol. Catal. B: Enzym., 2001, 15, 1–8 CrossRef CAS.
- J. F. B. Pereira, F. Vicente, V. C. Santos-Ebinuma, J. M. Araújo, A. Pessoa, M. G. Freire and J. A. P. Coutinho, Process Biochem., 2013, 48, 716–722 CrossRef CAS.
- S. P. M. Ventura, R. L. F. de Barros, J. M. de Pinho Barbosa, C. M. F. Soares, A. S. Lima and J. A. P. Coutinho, Green Chem., 2012, 14, 734–740 RSC.
- C. M. S. S. Neves, J. F. O. Granjo, M. G. Freire, A. Robertson, N. M. C. Oliveira and J. A. P. Coutinho, Green Chem., 2011, 13, 1517–1526 RSC.
- A. F. M. Cláudio, A. M. Ferreira, C. S. R. Freire, A. J. D. Silvestre, M. G. Freire and J. A. P. Coutinho, Sep. Purif. Technol., 2012, 97, 142–149 CrossRef.
- N. C. Stellwagen, A. Bossi, C. Gelfi and P. G. Righetti, Anal. Biochem., 2000, 287, 167–175 CrossRef CAS PubMed.
- M. R. Eftink and M. C. R. Shastry, Methods Enzymol., 1997, 278, 258–286 CAS.
- H. Morawetz, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1725–1735 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra16317j |
| This journal is © The Royal Society of Chemistry 2015 |