
Chromogenic naked-eye detection of copper ion and fluoride†
Abstract
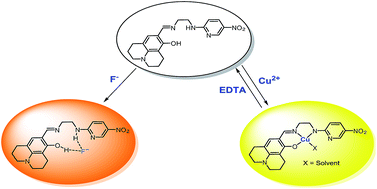
A bi-functional colorimetric chemosensor 1, based on julolidine moiety and N-(2-aminoethyl)-5-nitropyridin-2-amine, has been synthesized and characterized. The sensor 1 has proven to be highly selective and sensitive to Cu2+ with a color change from colorless to yellow in aqueous solution. The sensing mechanism of 1 for Cu2+ was proposed to be the ligand-to-metal charge-transfer (LMCT), which was explained by theoretical calculations. It was also found that the 1–Cu2+ complex could be recycled simply through treatment with an appropriate reagent such as EDTA. Importantly, the sensor 1 could be used to detect and quantify Cu2+ in water samples. Moreover, 1 showed a selective colorimetric response toward fluoride due to the increase in the intramolecular charge transfer (ICT) band by a deprotonation process without any inhibition from other anions such as CH3COO− and CN−.
 Please wait while we load your content...
Please wait while we load your content...

