
A facile and highly sensitive impedimetric DNA biosensor with ultralow background response based on in situ reduced graphene oxide
Abstract
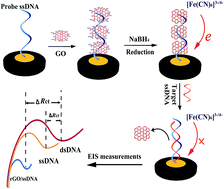
A novel electrochemical impedimetric DNA sensor was constructed based on in situ chemical reduction of graphene oxide (GO) that had been attached at a DNA modified electrode. First, the mercapto-modified probe DNA was anchored on a gold electrode surface through the Au–S bond. Then the GO was adsorbed on the probe DNA through the unique π–π stacking, which was followed by incubation in sodium borohydride (NaBH4) solution to in situ reduce the GO to the reduced form (rGO). Thus, a highly conductive biointerface with ultralow charge-transfer resistance was obtained. When the biosensor was hybridized with the target DNA to form the rigid double-stranded DNA, the rGO was released from the electrode surface and the charge-transfer resistance increased again. Compared with the analogous sensing interface without pre-accumulation of GO, the signal variation ratio was found to increase by 8-fold upon hybridization as determined by electrochemical impedance spectra, suggesting a higher signal-to-noise of the constructed biosensor. Quantitative analysis experiments showed that the impedance change values exhibited a good linear relationship with the logarithmic values of target DNA concentration over a wide range from 1.0 × 10−15 M to 1.0 × 10−9 M. The detection limit was estimated to be as low as 2.9 × 10−16 M. The biosensor also presented excellent selectivity, good regeneration ability and outstanding stability.
 Please wait while we load your content...
Please wait while we load your content...

