
On the investigation of hybrid quinones: synthesis, electrochemical studies and evaluation of trypanocidal activity†

Abstract
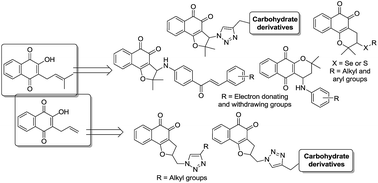
In our continued search for novel trypanocidal compounds, arylamine, chalcone, triazolic, triazole–carbohydrate and chalcogenium derivatives containing a naphthoquinone scaffold were prepared; in addition to electrochemical studies, these compounds were evaluated against the infective bloodstream form of Trypanosoma cruzi, the etiological agent of Chagas disease. Among the thirty-eight compounds herein evaluated, six were found to be more potent against trypomastigotes than the standard drug benznidazole, with IC50/24 h values between 52.9 and 89.5 μM.
 Please wait while we load your content...
Please wait while we load your content...

