
Synthesis, characterization, and cure chemistry of high performance phosphate cyanate ester resins†
Abstract
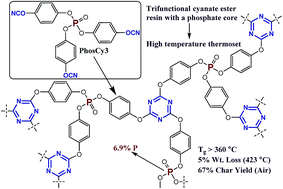
Three thermosetting cyanate ester resins with phosphate cores have been synthesized from chlorophosphates by a straightforward, three-step approach. A p-substituted bis(cyanate) ester generated by this route (PhosCy) was cured to give a thermoset material with a Tg of 223 °C and a char yield of 47% in air, while the m-substituted version (MPhosCy) yielded a thermoset with a Tg of only 131 °C and a char yield of 65% in air. The low Tg of the MPhosCy thermoset is attributed to a unique intramolecular cyclization of the cyanate ester groups, while the high char yield is attributed to a high temperature cross-linking reaction involving the phosphate core. To further explore this class of materials, a trifunctional phosphate cyanate ester (PhosCy3) was prepared from POCl3. This material was cured to generate a thermoset with an impressive Tg of >360 °C and a char yield of 67% in air. All three hybrid resins can potentially be used in fire-resistant composite materials or as protective surface coatings for conventional polymers.
 Please wait while we load your content...
Please wait while we load your content...

