
One-shot carboxylation of microcrystalline cellulose in the presence of nitroxyl radicals and sodium periodate
Abstract
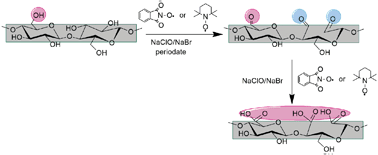
Water soluble cellulose derivatives are highly required products for many practical purposes, expanding the limited applications of pure cellulose caused by the highly ordered hydrogen bond network and high crystallinity. In this connection, this paper, presents a new approach to obtain water soluble carboxyl-functionalized cellulosic materials, combining two of the most common selective oxidation protocols for cellulose, i.e. nitroxyl mediated reaction and periodate oxidation, in a one-shot reaction. It was found that, under specific reaction conditions, fully oxidized, 2,3,6-tricarboxy cellulose can be obtained in large amounts. The other valuable oxidized fractions were found to possess large amounts of carboxylic groups, as determined by potentiometric titration. 13C-NMR evidenced the presence of three distinctive carboxylic groups in the fully oxidized product, whereas for the partially oxidized samples, 13C CP-MAS solid-state NMR did not detect any carbonyl signals. The oxidized products were characterized by means of FTIR and X-ray photoelectron spectroscopy (XPS). Moreover, the changes of the degree of polymerization occurring after oxidative treatments were viscometrically determined.
 Please wait while we load your content...
Please wait while we load your content...

