
Synthesis of silicone elastomers containing trifluoropropyl groups and their use in dielectric elastomer transducers†
Mihaela Dascalu‡
a,
Simon J. Dünkiab,
Jose-Enrico Q. Quinsaatab,
Yee Song Koab and
Dorina M. Opris*a
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Functional Polymers, Ueberlandstr. 129, CH-8600, Dübendorf, Switzerland. E-mail: dorina.opris@empa.ch
bEcole Polytechnique Fédérale de Lausanne (EPFL), Institut des matériaux, Station 12, CH 1015, Lausanne, Switzerland
First published on 1st December 2015
Abstract
Vinyl end-functionalized polysiloxanes Px containing varying mol% of trifluoropropyl groups (x) were prepared starting from 1,3,5-tris(3,3,3-trifluoropropyl)-1,3,5-trimethylcyclotrisiloxane (F3) and octamethylcyclotetrasiloxane (D4) via anionic polymerization in the presence of tetramethylammonium hydroxide (TMAH) and 1,3-divinyl-1,1,3,3-tetramethyldisiloxane end-capping reagent. Their structures were determined by 1H NMR spectroscopy and their molecular weights and distributions were measured by GPC. The various Px were cross-linked in thin films via hydrosilylation of the vinyl groups with tetrakis(dimethylsiloxy)silane cross-linker in the presence of Karstedt catalyst. The mechanical, dielectric and electromechanical properties of the prepared films were investigated. An increase in the permittivity (ε′) with increasing content of polar trifluoropropyl groups was observed with a maximum value of ε′ = 6.4 for P58(0). A maximum lateral actuation strain of 5.4% at an electric field as low as 7.8 V μm−1 was measured for a material prepared by cross-linking P53.
Introduction
Cross-linked polydimethylsiloxanes (silicones) are presently intensively explored as dielectrics in dielectric elastomer transducers (DET).1,2 DET are stretchable capacitors that elongate when charged and can function as actuators, generators, and sensors.3 A large variety of applications have been proposed, which range from valves, pumps, wave generators, pressure sensors, to muscle replacements, to name a few.4Any elastomer can in principle be used as a dielectric in DET, however silicones are most often used mainly due to their fast response time, low viscoelastic loss, and low Tg which allow operation at different frequencies and temperatures. Silicones have a high dielectric strength, high gas permeability and hydrophobicity which afford them with humidity tolerance and a low water uptake.5 Silicone actuators can have large actuation strains and high energy densities, but rather high electric fields have to be used.6,7 More efficient silicones that actuate at low electric fields might pave the way for application of this technology in the area of medicine, e.g. as implantable devices, where the maximum allowed operation voltage is below 24 V.8 The actuation strain can be increased when the thickness of the dielectric film is reduced and when soft high permittivity elastomers are used as dielectric. Another possibility to enhance the actuation strain is to use appropriately designed heterogeneous materials consisting of alternating soft isotropic and anisotropic layers. For such materials theoretical calculations predict a ten-fold enhancement of the electromechanical coupling.9
The ε′ of polymers can be increased when blended with high permittivity ceramic10,11 or conductive particles.12–17 As fillers, polyaniline,12 encapsulated polyaniline in polydivinylbenzene,13 silver nanoparticles coated with a thin silica shell,14 carbon black,15 carbon nanotubes,16 and polythiophene17 have been used. An increase in permittivity was observed with increasing volume fraction of conductive filler, but other properties like elasticity, elastic moduli, strain at break, dielectric loss, and dielectric breakdown were negatively affected. Furthermore, the migration of the conductive filler in an electric field and thus the long term stability of such composites are of concern especially when thermoplastic matrices are used. Ceramic fillers significantly increase the permittivity, but concentrations above 50 vol% have to be used. The resulting materials tend to have poor mechanical properties and increased elastic moduli. The present work is focused on increasing the electromechanical response in an actuator by using soft high permittivity silicones.
Silicones have a rather low ε′ of less than 3 and therefore high electric fields are required to induce actuation. It has been suggested that the ε′ of polymers can be increased when modified with polar groups.18 When modified with nitroaniline and nitrobenzene moieties, silicones show ε′ = 6 and ε′ = 8.5 at high frequencies, respectively,19,20 while ε′ = 4.7 can be achieved when modified with chloropropyl groups.21 By modifying the silicones with cyanopropyl groups at every repeat unit, polymers with ε′ of about 18 were prepared.22,23 Recently, the synthesis of a silicone elastomer modified with polar nitrile groups was reported which combines ε′ of about 10, low elastic moduli and a strain at break of 260%.24 Commercial fluorinated silicones also show a high permittivity and were used by Pelrine et al. as dielectric in actuators.25 Unfortunately, the reported materials are commercial products and therefore no data about their chemical composition is available. More recently, Böse et al. used a trifluoropropyl (CF3) modified silicone oil as softening agent in a polydimethylsiloxane matrix and obtained a material with ε′ = 5.5, a strain at break as high as 400%, and a maximum actuation strain of 7.5% at 14 V μm−1.26 The polar CF3 modified silicone oil was mixed together with a nonpolar polydimethylsiloxane and thus the resulting materials might undergo phase separation. Furthermore, the amount of CF3 modified silicone used in the silicone matrix was limited to 45% which represents about 28 mol% of CF3 groups in the material. A further increase of the CF3 content resulted in very soft materials which were difficult to process in thin films.
The aim of this work was to synthesize high molecular weight polysiloxanes Px containing different mol% of CF3 groups as well as reactive end-groups which allow them to be cross-linked in thin films. Homogenous materials were prepared, for which the dielectric, mechanical, and electromechanical properties were investigated as function of CF3 content.
Experimental section
Materials and characterization
1,3,5-Tris(3,3,3-trifluoropropylmethyl)-1,3,5-trimethylcyclotrisiloxane (F3) was purchased from EuroLab Limited. Octamethylcyclotetrasiloxane (D4) and tetrakis(dimethylsiloxy)silane were purchased from ABCR, 1,3-divinyl-1,1,3,3-tetramethyldisiloxane (EB), TMAH (25 wt% solution in methanol) and platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex solution in xylene (Pt ∼ 2 wt%) were purchased from Sigma-Aldrich. Prior to use D4 and F3 were dried over calcium hydride, and TMAH was dried by azeotropic distillation. All other chemicals were used as received. Acrylic foil VHB-4905 from 3M was used.FTIR spectra were recorded on a Bruker Vertex 70 ATR FT-IR spectrometer at room temperature. 1H NMR spectra were recorded on a Bruker Avance III 400 NMR spectrometer using a 5 mm BBO Prodigy™ CryoProbe at 400.18 Hz. GPC was conducted using an Agilent 1100 Series HPLC (columns: serial coupled PSS SDV 5 u, 100 A and PSS SDV 5 u, 1000 A, detector: DAD, 235 nm and 360 nm; refractive index) with THF solvent using polydimethylsiloxane and toluene as internal standards. Thermogravimetric analysis (TGA) was conducted on a Perkin Elmer TGA7 at a heating rate of 20 °C min−1 under a nitrogen gas flow up to 800 °C. Differential scanning calorimetry (DSC) measurements were conducted on a Perkin Elmer DSC 8000. The mechanical properties were investigated with a Zwick Z010 tensile test machine with a crosshead speed of 200 mm min−1 (strain rate of 1111.1% min−1). Tensile test specimens with a gauge width of 2 mm and a gauge length of 18 mm were prepared by die cutting. The strain was determined using a traverse moving sensor. The curves were averaged from 2–4 different specimens (see ESI†). The tensile modulus was determined from the slope of the stress–strain curves using a linear fit to the data points within 10% strain. The strain at break was calculated by taking the average of the different measurements. DMA spectra were measured on a ARES Rheometer from TA instruments with a parallel plate setup (25 mm plate diameter), a strain of 5% and 1 N preforce. Dielectric measurements were conducted on a Novocontrol Alpha-A Frequency Analyzer using a Hewlett Packard 16451B Dielectric test fixture as electrodes. Shielded electrodes with a diameter of 5 mm were used and the sample thickness was determined by using the built in micrometer screw. The probing voltage was 1RMSV (root mean square voltage). Actuator tests were performed using circular membrane actuators, for which the films were fixed between two circular frames of 25 mm diameter. Circular electrodes (8 mm diameter) of carbon black powder were applied to each side of the film. A high voltage amplifier Trek Model 5/80 (up to 5600 V) was used for actuator tests. The voltage was increased by 100 V every 2 s. The actuation strain was measured optically as the extension of the diameter of the electrode area via a digital camera, using an edge detection tool of a LabView program to detect the boundary between the black electrode area and brighter film.
General procedure for the synthesis of Px
A solution of TMAH in methanol (73 μL) was added to a three-neck 50 mL round-bottom flask and dried by azeotropic distillation. D4, F3 and EB were added simultaneously to the dried TMAH. For the amount of reagents used, see Table 1. The reaction mixture was heated to 100 ± 2 °C under nitrogen for 4.5 h. Then the reaction mixture was heated to 160 °C and kept at this temperature for 2 h to deactivate the initiator. Finally, the unreacted cycles were distilled at 180–200 °C under vacuum. The residue was washed with ethanol and then dried in an oven at 100 °C to obtain Px. 1H NMR (400 MHz, CDCl3, δ): 6.20–5.71 (vinyl–H), 2.08 (CH2– –CF3), 0.76 (
–CF3), 0.76 ( –CH2–CF3), 0.1 (Si–CH3). The chemical composition of Px as well as its molar mass and distribution are given in Table 2.
–CH2–CF3), 0.1 (Si–CH3). The chemical composition of Px as well as its molar mass and distribution are given in Table 2.
| Entry | F3 g (mmol) | D4 g (mmol) | F3/D4 mol ratio | EB μL (mmol) | TMAHa [μL] |
|---|---|---|---|---|---|
| a A solution of 25% in methanol was used. | |||||
| P28 | 11.2 (23.9) | 7.1 (23.9) | 1 | 23 (0.1) | 73 |
| P42 | 22.3 (47.6) | 14.2 (47.9) | 1 | 45 (0.2) | 128 |
| P47 | 21.7 (46.3) | 10.6 (35.7) | 1.3 | 45 (0.2) | 128 |
| P53 | 21.7 (46.3) | 9.2 (31.0) | 1.5 | 45 (0.2) | 123 |
| P58 | 21.7 (46.3) | 5.9 (19.9) | 2.3 | 45 (0.2) | 110 |
| Entry | NMR | GPC | ||||
|---|---|---|---|---|---|---|
| Fa [mol%] | Db [mol%] | Mn [g mol−1] | Mn [g mol−1] | Mw [g mol−1] | PDI | |
| a F represents trifluoropropyl.b Represents dimethylsilyl. | ||||||
| P28 | 28.3 | 71.7 | 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
46800 |
101700 |
2.2 |
| P42 | 41.8 | 58.2 | 118500 |
59520 |
140200 |
2.4 |
| P47 | 47.1 | 52.9 | 13400 |
11600 |
18200 |
1.6 |
| P53 | 52.9 | 47.1 | 48500 |
34700 |
75900 |
2.2 |
| P58 | 57.5 | 42.5 | 47000 |
16500 |
48900 |
3.0 |
General synthesis of polysiloxane elastomers containing trifluoropropyl groups
To a homogenous mixture of Px and tetrakis(dimethylsiloxy)silane cross-linker (CL, 10 wt% in toluene), a solution of Karstedt catalyst (1 vol% in toluene) was added. This mixture was stirred for another 3 min and sonicated for 1 min to remove the air bubbles. To reinforce the elastomers, a dispersion of hexamethyldisilasane (HMDS) treated silica particles in toluene was used. Thin films were prepared by the doctor blade technique. The samples were aged at 80 °C in oven for 24 h. The resulting elastomers were named as: Px(y), where Px represents the starting copolymer and y represents the wt% of silica used. Table 3 summarises the amounts of reagents used for the synthesis of Px(y).| Sample | Px [g] | CLa [μL] | Molar ratio [vinyl/Si–H] | Karstedt cat.b [μL] | SiO2d [mg] |
|---|---|---|---|---|---|
| a A 10 wt% solution of tetrakis(dimethylsiloxy)silane in toluene was used.b 1 vol%.c 0.5 vol% of Karstedt catalyst in toluene was used.d Silica particles were dispersed in toluene prior use. | |||||
| P28(0) | 0.5 | 53 | 1.1 | 32.4 | — |
| P28(5) | 0.5 | 53 | 1.1 | 32.4 | 25 |
| P28(10) | 0.5 | 53 | 1.1 | 32.4 | 50 |
| P42(0) | 2.0 | 111 | 1.1 | 67.5 | — |
| P42(5) | 2.0 | 111 | 1.1 | 67.5 | 100 |
| P42(10) | 2.0 | 111 | 1.1 | 67.5 | 200 |
| P47(0) | 0.5 | 62 | 4.5 | 18.7c | — |
| P47(5) | 0.5 | 62 | 4.5 | 18.7c | 25 |
| P47(10) | 0.5 | 62 | 4.5 | 18.7c | 50 |
| P53(0) | 2.0 | 271 | 1.1 | 164.8 | — |
| P53(5) | 2.0 | 271 | 1.1 | 164.8 | 100 |
| P57(0) | 2.0 | 279 | 1.1 | 170.0 | — |
| P57(5) | 2.0 | 279 | 1.1 | 170.0 | 100 |
Results and discussions
Synthesis and characterization of Px
Vinyl end-functionalized polysiloxanes containing varying mol% of CF3 groups Px were prepared starting from F3 and D4 via anionic bulk polymerization in the presence of TMAH catalyst and 1,3-divinyl-1,1,3,3-tetramethyldisiloxane end-capping reagent (Scheme 1). TMAH was selected as initiator because it can be decomposed to inactive components at elevated temperatures, while the end capping reagent allowed us to functionalize Px with vinyl end-groups that should be further used for the cross-linking in thin films. The content of CF3 groups in Px was tuned by using different feed ratios of D4 to F3, while the molecular weight can be controlled by the amount of end-capping reagent used. Px were analysed by 1H NMR, IR and GPC.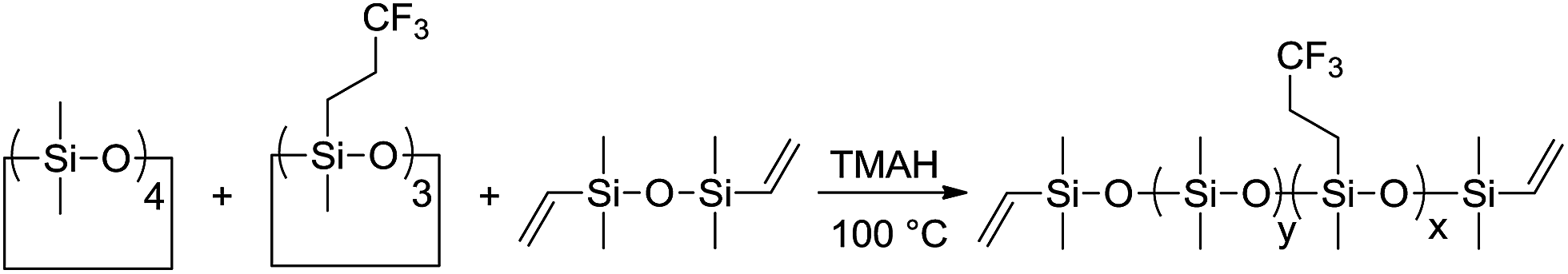 | ||
| Scheme 1 Synthesis of vinyl end-functionalized polysiloxanes containing CF3 groups starting from D4, F3, and 1,3-divinyl-1,1,3,3-tetramethyldisiloxane end-blocker. | ||
Fig. 1 shows the 1H NMR spectra of P47. The signal at δ = 0.10 ppm is assigned to the methyl groups, the signals at δ = 0.76 ppm and δ = 2.08 ppm correspond to the methylene units α- and β- to CF3 groups.27 The signals between δ = 5.71 to δ = 6.20 ppm are attributed to the vinyl end-groups.28 The other copolymers Px show the same signals as those of P47, but the intensities of the signals are different (Fig. S1–S5†). The content of CF3 groups in Px was calculated using the integrals of the signal at δ = 0.10 ppm and that at δ = 2.08 of the methylene units α to CF3. Although F3 has a higher reactivity as compared to D4, the amount of CF3 units incorporated in the polymers was found to be lower than the feed ratios F3:D4. The reason for this might be the high temperature used for the decomposition of TMAH which might favour the depolymerization and formation of cyclic monomers containing CF3 units. Nevertheless, an increase in content of CF3 units in Px with increasing F3:D4 feed ratios was observed.
 | ||
| Fig. 1 1H NMR spectrum of P47 in CDCl3 at room temperature. | ||
Mn values were calculated by comparing the integral of the signal at δ = 0.10 ppm with the integral of the vinyl protons and found to be slightly higher than the Mn values obtained from GPC (Fig. S6–S10†).29 Table 2 gives an overview of the chemical composition of Px as well as the molecular weight and its distribution as found by NMR (ESI Fig. S1–S5†) and GPC measurements (see ESI†).
The infrared absorption spectra of Px are shown in Fig. S11 (see ESI†). The broad and strong absorption band from 1170 cm−1 to 1000 cm−1 in each spectrum is attributed to the Si–O–Si asymmetric stretching vibration.30,31 The absorption bands from 2964 cm−1 and 2909 cm−1 are assigned to the C–H vibrations from –CH3 groups. The peak at 1264 cm−1 is assigned to the absorption due to vibration of Si–CH3. Characteristic absorption peaks of –CH2CH2CF3 appeared at 1368 cm−1 (wagging –CH2–), 1315 cm−1 (CH2–CH2), 1208 cm−1 (–CF3), 1168 cm−1 (C–H bond in –CH2–) and 902 cm−1 (C–CF3). An absorption band in the region of 740–850 cm−1, originating from –CH3 rocking and Si–C stretching is also clearly visible. This absorption peak became broader and the peak height decreased gradually with increasing mol% of CF3 groups. Additionally, the main absorption peak of Si–C bond shifted to a slightly higher frequency, as did the absorption peak of C–H bonds in –CH3, which further supports the presence of the CF3 units. Furthermore, all the absorption intensities of the characteristic peaks of CF3 increased with increasing feed ratio of F3, which suggested that the content of CF3 groups in Px increased along with increasing F3 feed ratio. The presence of vinyl groups is confirmed by a broad and weak absorption band from 1600 cm−1 to 1720 cm−1 present in each spectrum.32 As mentioned above, the end-functionalization of Px with vinyl groups is important, since these groups are used to cross-link Px in thin films.
The thermal stability of Px was investigated by TGA in argon atmosphere and shows that Px are stable up to 380 °C and have a maximum weight loss around 450 °C (Fig. 2).
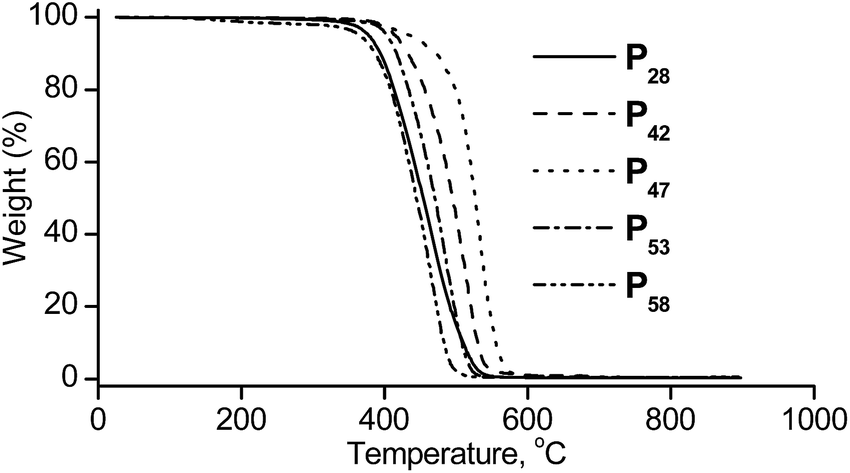 | ||
| Fig. 2 TGA curves of Px conducted at a heating rate of 20 °C min−1 under nitrogen. | ||
Differential scanning calorimetry (DSC) measurements show an increase in Tg with increasing mol% of CF3 groups from −106 °C for P28 to −88.6 °C for P58 (ESI Fig. S12–S16,† Table 4). It is known that the introduction of polar moieties in a polymer shifts the Tg of the modified polymer to higher values. Luckily, the Tgs of Px are well below room temperature and thus the materials obtained by cross-linking Px are still elastic. The presence of only one glass transition temperature indicates that the distribution of CF3 groups in Px is random. No melting temperatures were observed in the DSC. PDMS is known to easily crystallize at about −80 °C.33 However, by modifying the silicone with CF3 group, the crystallization process in Px is hindered. Even for P28, which has the lowest content of CF3 groups, no melting temperature was observed.34
| Copolymer | Tg [°C] | ΔCp [J g−1 °C−1] | ||
|---|---|---|---|---|
| 2nd heating | Cooling | 2nd heating | Cooling | |
| P28 | −106.0 | −115.4 | 0.39 | 0.47 |
| P42 | −97.7 | −105.8 | 0.27 | 0.28 |
| P47 | −97.1 | −103.6 | 0.30 | 0.25 |
| P53 | −92.2 | −98.0 | 0.33 | 0.24 |
| P58 | −88.6 | −95.8 | 0.31 | 0.25 |
Synthesis and characterization of Px(y)
All materials of series Px are highly viscous liquids and have to be cross-linked to furnish elastomers. As mentioned before, we designed Px with vinyl end-groups which should allow cross-linking. Next, the vinyl end-functionalized Px were cross-linked via hydrosilylation using a tetrafunctional cross-linker tetrakis(dimethylsiloxy)silane and Karstedt's platinum catalyst. The molar ratio of the vinyl groups of Px to the hydrosilyl groups of the CL was kept constant (1.1) for all synthesized materials except for P47 for which the ratio was increased. The reason for this was the fast cross-linking observed when P47 was cross-linked, which did not allow sufficient time for thin film preparation. A reduction of the catalyst did not reduce the reaction rate significantly. Therefore the amount of cross-linker used had to be decreased. To optimize the mechanical properties of the resulting materials, hexamethyldisilazane treated silica particles were used as reinforcing filler. Thin films were prepared by the doctor blade technique on a Teflon substrate. The synthesized materials were named as Px(y), where Px represents the copolymer and y represents the wt% of surface treated silica particles used.The mechanical properties of Px(y) were investigated in tensile tests (see ESI Fig. S17–S28†). Fig. 3 shows the stress–strain curves of materials Px(y), while Table 5 gives an overview of the Young's moduli at different strains (Yx%) and the strain at break. It is known that the mechanical properties of a material are strongly affected by the molecular weight of the polymer used. Unfortunately, because the molar masses of Px were rather different, a direct comparison of the mechanical properties as function of CF3 content was not possible. However, irrespective of the content of trifloropropyl units in the materials and the Mw of the polymer used, an increase in the tensile strength and a decrease in the elongation at break with increasing amount of silica were observed. Materials Px(y), for which the concentration of cross-linker to vinyl groups was kept constant, showed an increase in the strain at break with increasing molar mass of polymer Px used. The lowest strain at break of 180% was observed for material P47(10) which was synthesized starting from P47 and had the lowest Mw, while the maximum strain at break of 850% was observed for P42(0) which was prepared starting from P42 and had the highest Mw. All other materials showed an elongation at break value between 240 to 550%. It should be mentioned here that materials P42(y) showed visible viscous flow when strained, were sticky and difficult to handle in thin films. For materials P58(y) which have the highest CF3 content, the strain at break was slightly improved when 5 wt% silica was used and the tensile strength was increased from 0.05 MPa for P58(0) to 0.22 MPa for P58(5). Sample P53(5) showed an increased tensile strength, while the strain at break was only slightly affected as compared to P53(0). Furthermore, an increase in the stickiness with CF3 content was observed. All obtained materials Px(y) are rather soft and have Young's moduli (at 10% strain) that range between 19 kPa to 405 kPa. Additionally an increase in the Young's moduli with the addition of silica filler was observed (see Table 5).
 | ||
| Fig. 3 Stress–strain curves for Px(y) (left) with the stress–strain curves up to 200% strain enlarged (right). The strain at break represents the lowest value obtained from different measurements. Tensile test specimens with a gauge width of 2 mm and a gauge length of 18 mm were tested at a crosshead speed of 200 mm min−1. | ||
| Sample | Young modulusa [kPa] | Strain breakc [%] | |||
|---|---|---|---|---|---|
| 10% (0–20) | 50% (40–60) | 100% (90–110) | 200% (190–210) | ||
| a Young's moduli at different strain levels.b The slope between 5 and 15% strain was taken.c The average of the strain at break from different measurements is given. | |||||
| P28(0) | 58 | 26 | 22 | 21 | 513 |
| P28(5) | 65 | 35 | 34 | 44 | 488 |
| P28(10) | 97 | 42 | 46 | 61 | 552 |
| P42(0) | 24 | 13 | 11 | 10 | 850 |
| P42(5) | 45 | 26 | 22 | 27 | 540 |
| P47(0) | 50 | 30 | 24 | 25 | 360 |
| P47(5) | 184 | 107 | 104 | 150 | 240 |
| P47(10) | 405 | 298 | 352 | — | 180 |
| P53(0) | 19 | 15 | 15 | 13 | 498 |
| P53(5) | 72 | 46 | — | — | 459 |
| P58(0) | 20 | 17 | 18 | — | 263 |
| P58(5) | 34 | 28 | 34 | 64 | 349 |
| VHB | 146b | 50 | 30 | 27 | |
The dielectric properties of materials Px(y) were also measured in a frequency range from 10−1 Hz to 106 Hz and room temperature (Fig. 4). Table 6 summarizes ε′, dielectric loss (ε′′) and conductivity (σ) at 10 kHz for materials Px(0), Px(5) and VHB foil. ε′ raised with increasing amounts of CF3 groups in the materials. The ε′ values are almost constant at frequencies above 102 Hz. The increase in ε′ at low frequencies is most likely caused by electrode polarization.35 The ε′ at 10 kHz for Px(y) as well as for F3 monomer (ε′ = 8.8) are given as function of CF3 content in Fig. 5. A linear increase in ε′ with increasing amounts of CF3 groups from ε′ = 5.1 for P28(0) to ε′ = 6.4 for P58(0) was observed. Since the permittivity is increasing with the concentration of the polar groups in the material, it is expected that a material P100(0) would have a maximum ε′ = 8.8. Materials Px(5) and Px(10) which contain 5 and 10 wt% silica particles show slightly lower values for the permittivity as compared to Px(0). This decrease is not unexpected given the fact that silica has ε′ = 3.9. The conductivity at low frequencies (the static conductivity) is lower than 10−10 S cm−1, typical for insulating materials.36 The silica modified materials Px(5) and Px(10), tend to have a lower conductivity than Px(0) (Fig. 4).
 | ||
| Fig. 4 Dielectric properties as function of frequency of Px(y). | ||
| Sample | ε′a | ε′′a | σa [S cm−1] | Tanδ |
Y10% [kPa] | ε′/Y [MPa−1] | s [%] at 7.8 V μm−1 | smax [%] | Eb [V μm−1] | dd [μm] |
|---|---|---|---|---|---|---|---|---|---|---|
| a The permittivity, dielectric losses and conductivity values were taken at 10 kHz.b Maximum field achieved, no breakdown.c Y at 300% strain.d Actuator thickness. | ||||||||||
| P28(0) | 5.1 | 0.018 | 8.92 × 10−11 | 0.0035 | 58 | 88 | 1.1 | 4.8 | 19.1 | 89 |
| P28(5) | 4.5 | 0.005 | 2.95 × 10−11 | 0.0012 | 65 | 69 | — | — | — | — |
| P42(0) | 5.6 | 0.004 | 1.89 × 10−11 | 0.0007 | 24 | 233 | — | 1.8 | 6.5 | 175 |
| P42(5) | 5.3 | 0.004 | 2.40 × 10−11 | 0.0008 | 45 | 118 | 0.8 | 5.1 | 17.1 | 317 |
| P47(0) | 5.7 | 0.007 | 3.37 × 10−11 | 0.0012 | 50 | 114 | 0.9 | 3.7 | 16.0b | 317 |
| P47(5) | 5.7 | 0.004 | 2.03 × 10−11 | 0.0007 | 184 | 31 | — | — | — | — |
| P53(0) | 6.2 | 0.005 | 2.35 × 10−11 | 0.0008 | 19 | 326 | 5.4 | 5.4 | 7.8 | 320 |
| P53(5) | 6.0 | 0.005 | 2.43 × 10−11 | 0.0009 | 72 | 83 | 1.1 | 3.6 | 13.3 | 223 |
| P58(0) | 6.4 | 0.007 | 3.37 × 10−11 | 0.00011 | 20 | 320 | — | — | — | — |
| P58(5) | 6.1 | 0.005 | 2.66 × 10−11 | 0.0009 | 34 | 179 | 3.1 | 5.0 | 10.2 | 157 |
| VHB | 4.4 | — | 9.02 × 10−10 | 0.044 | 32c | 138 | — | — | >100 | — |
 | ||
| Fig. 5 Dielectric permittivity at 10 kHz of Px(y) as function of mol% of CF3 groups. The permittivity at 100% is the permittivity of monomer F3. | ||
In all materials Px(y), a minimum in the dielectric losses of less than 10−2 was observed between 104 and 105 Hz which recommends these materials for high frequency applications.
Some materials were further investigated in electromechanical tests using circular actuators. The voltage was increased in steps of 100 V every 2 s until breakdown through the material occurred. Fig. 6 shows the lateral actuation strain as a function of the electric field, while Table 6 summarizes the maximum actuation strain achieved at breakdown, and the actuation strains at 7.8 V μm−1 for Px(y). All tested materials showed actuation at rather low electric fields of less than 20 V μm−1. The best performance in terms of actuation strain at a low electric field was observed for material P53(0) which has the highest ratio of ε′/Y and showed an actuation strain of 5.4% at 7.8 V μm−1, while material P53(5) showed significantly lower actuation strain of 1.1%. The reason behind this is its slightly lower permittivity value and higher Young's moduli. Material P58(5) showed slightly lower actuation strain as compared to P53(0) most likely due to its higher elastic modulus. The lowest actuation strain was observed for material P28(0) which had the lowest content of CF3.
 | ||
| Fig. 6 Lateral actuation strain of circular actuators of materials Px(y), as a function of applied electric field. The voltage was increased by 100 V every 2 s (0.5 Hz). The actuator constructed from P47(0) did not suffer a breakdown at 16 V μm−1, however, we could not increase the voltage further due to the limitation of the voltage source used. | ||
A raise in the actuation strain at a certain electric field with increasing content of CF3 groups was observed for materials Px(5). For example, materials P42(5) and P58(5) showed about the same actuation strain of 5% but the electric field used decreased from 17.1 V μm−1 for P42(5) to 10.2 V μm−1 for P58(5), respectively. Material P28(0) shows the highest breakdown field of 19.1 V μm−1. The film prepared from material P47(0) was rather thick (317 μm) and therefore, the maximum given actuation is not the actuation at breakdown. Furthermore, a decrease in actuator breakdown field with increasing content of polar component from 19.1 V μm−1 for P28(0) to 7.8 V μm−1 for P53(0) was observed.
As mentioned above, materials P42(y) after straining, do not immediately recover their initial shape. Room temperature rheological measurements show that the storage moduli G′ for the tested material are rather low, typical for elastomeric materials (see DMA in ESI Fig. S29†). The viscoelastic losses of different materials negatively affect the cyclic actuation strain at a certain frequency (see ESI, Fig. S30–S33†).
For example, while the cyclic actuation of P47(0) at 17.6 V μm−1 showed only a small hysteresis in the actuation strain in time (Fig. 7 and S34†), materials P42(y) showed quite some hysteresis and require a few cycles to achieve a stable actuation (see ESI, Fig. S31†). Cyclic actuation tests for materials P53(y) and P58(5) also show a rather small hysteresis (see ESI, Fig. S35–S37†). Some actuators, especially those constructed from materials with high CF3 content showed a repairing effect which was observed during the cyclic actuation tests (see ESI, Fig. S36†). Although this effect is of potential advantage since the lifetime and the reliability of the actuator is increased, this process was rather slow and can occasionally take minutes.
 | ||
| Fig. 7 Long-term stability of P47(0) actuator at 17.6 V μm−1 at 28.6% prestrain (100 cycles at 0.4 Hz). | ||
Finally, the performance of our material will be compared to other reported materials26 and VHB. An elastomer containing 45 wt% CF3 modified silicone which represents about 28 mol% of CF3 units in the final material has a similar chemical composition as P28(0). The reported material has a ε′ = 5.4, which is in agreement with the values measured by us (ε′ = 5.1). While the reported material has a strain at break of 280%, P28(0) shows a much higher strain at break of 513%. Unfortunately, the actuation strain of the two materials cannot be easily compared, since different setups were used. The VHB foil is widely used by the DEA community and it was therefore selected here as reference material. The mechanical and dielectric characteristics of VHB were included in Tables 5 and 6.37 The VHB membrane was prestrained biaxially by 300% before circular carbon black electrodes with a diameter of 8 mm were applied. As can be seen in Fig. 6, by far the lowest actuation strain at a certain electric field is observed for VHB which at 20 V μm−1 showed only a 0.35% lateral actuation strain. However, in terms of maximum achievable actuation strain and dielectric breakdown, the VHB foil is still unbeatable. However, for implantable devices for which the maximum allowed voltage is 24 V, the materials presented herein are superior. Future work will focus on the upscaling of the synthesis and the optimization of the mechanical properties of materials P53(y) and P58(y) as well as on investigating how the dielectric breakdown of these materials can be increased.
Conclusions
Polysiloxanes Px containing different mol% of trifluoropropyl groups and vinyl end-groups were successfully obtained under anionic conditions starting from F3, D4, and 1,3-divinyl-1,1,3,3-tetramethyldisiloxane end-capping reagent. By cross-linking Px in thin films using a hydrosilylation reaction, elastomers with good mechanical and dielectric properties were obtained. All the materials show a strain at break higher than 180%. The highest strain at break value was 850% and was obtained for a material for which a high molecular weight P42(0) was used. The dielectric permittivity raised with increasing amounts of polar trifluoropropyl groups and a maximum value of ε′ = 6.4 was measured for P58(0). An actuation strain of 5.4% at 7.8 V μm−1 where electrical breakdown occurred was measured for a film prepared starting from P53(0). The increased permittivity and the low actuation voltage recommend these materials as dielectrics in sensors and actuators.Acknowledgements
We gratefully acknowledge Sciex (project no. 12.192), the Swiss National Science Foundation (IZERZ0 – 142215/1 and No. 150638), and the Swiss Federal Laboratories for Materials Science and Technology for financial support. We also like to acknowledge B. Fischer (Empa, Switzerland) for the TGA and DSC measurements, D. Rentsch (Empa, Switzerland) for helping with the NMR measurements, and G. Kovacs (Empa, Switzerland) for providing us with the infrastructure for the actuator measurements. DMO thanks C. Racles from Petru Poni Institute, Iasi, Romania for her kind support of the Sciex fellow and Prof. F. Nüesch from Empa for his generous support.Notes and references
- P. Brochu and Q. Pei, Macromol. Rapid Commun., 2010, 31, 10–36 CrossRef CAS PubMed.
- J. Biggs, K. Danielmeier, J. Hitzbleck, J. Krause, T. Kridl, S. Nowak, E. Orselli, X. Quan, D. Schapeler, W. Sutherland and J. Wagner, Angew. Chem., Int. Ed., 2013, 52, 9409–9421 CrossRef CAS PubMed.
- R. Perline, R. Kornbluh, Q. Pei and J. Joseph, Science, 2000, 287, 836–839 CrossRef.
- F. Carpi, D. de Rossi, R. Kornbluh, R. Pelrine and P. Sommer-Larsen, in Dielectric Elastomers as Electromechanical Transducers: Fundamentals, Materials, Devices, Models and Applications of an Emerging Electroactive Polymer Technology, Elsevier, Oxford, 2011 Search PubMed.
- M. Iacob, A. Bele, X. Patras, S. Pasca, M. Butnaru, M. Alexandru, D. Ovezea and M. Cazacu, Mater. Sci. Eng., 2014, 43, 392–402 CrossRef CAS PubMed.
- S. Akbari and H. R. Shea, Sens. Actuators, A, 2012, 186, 236–241 CrossRef CAS.
- C. Racles, M. Cazacu, B. Fischer and D. M. Opris, Smart Mater. Struct., 2013, 22, 104004 CrossRef.
- B. Müller, H. Deyhle, S. Mushkolaj and M. Wieland, Swiss Med. Wkly., 2009, 139, 591 Search PubMed.
- S. Rudykh, A. Lewinstein, G. Uner and G. deBotton, Appl. Phys. Lett., 2013, 102, 151905 CrossRef.
- G. Gallone, F. Carpi, F. Galantini, D. de Rossi and G. Levita, Adv. Sci. Tech., 2008, 61, 46–52 CrossRef CAS.
- H. Stoyanov, M. Kollosche, S. Risse, D. N. McCarthy and G. Kofod, Soft Matter, 2011, 7, 194 RSC.
- C. Huang, Q. M. Zhang, G. deBotton and K. Bhattacharya, Appl. Phys. Lett., 2004, 84, 4391 CrossRef CAS.
- M. Molberg, D. Crespy, P. Rupper, F. Nüesch, J.-A. E. Månson, C. Löwe and D. M. Opris, Adv. Funct. Mater., 2010, 20, 3280–3291 CrossRef CAS.
- J. E. Q. Quinsaat, M. Alexandru, F. A. Nüesch, H. Hofmann, A. Borgschulte and D. M. Opris, J. Mater. Chem. A, 2015, 3, 14675–14685 CAS.
- H. Stoyanov, D. McCarthy, M. Kollosche and G. Kofod, Appl. Phys. Lett., 2009, 94, 202901 CrossRef.
- T. P. Selvin, A. A. Abdullateef, M. A. Al-Harthi, M. A. Atieh, S. K. De, M. Rahaman, T. K. Chaki, D. Khastgir and S. Bandyopadhyay, J. Mater. Sci., 2012, 47, 3344–3349 CrossRef.
- F. Carpi, G. Gallone, F. Galantini and D. de Rossi, Adv. Funct. Mater., 2008, 18, 235 CrossRef CAS.
- W. J. Feast, M. Gimeno and E. Khosravi, Polymer, 2003, 44, 6111–6121 CrossRef CAS.
- F. B. Madsen, I. Dimitrov, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polym. Chem., 2013, 4, 1700–1707 RSC.
- B. Kussmaul, S. Risse, G. Kofod, R. Waché, M. Wegener, D. N. McCarthy, H. Krüger and R. Gerhard, Adv. Funct. Mater., 2011, 21, 4589–4594 CrossRef CAS.
- F. B. Madsen, L. Yu, A. E. Daugaard, S. Hvilsted and A. L. Skov, RSC Adv., 2015, 5, 10254–10259 RSC.
- S. J. Dünki, M. Tress, F. Kremer, S. Y. Ko, F. A. Nüesch, C.-D. Varganici, C. Racles and D. M. Opris, RSC Adv., 2015, 5, 50054–50062 RSC.
- C. Racles, M. Alexandru, A. Bele, V. E. Musteata, M. Cazacu and D. M. Opris, RSC Adv., 2014, 4, 37620–37628 RSC.
- S. J. Dünki, Y. S. Ko, F. A. Nüesch and D. M. Opris, Adv. Funct. Mater., 2015, 25, 2467–2475 CrossRef.
- R. Pelrine, R. Kornbluh, J. Joseph, R. Heydt, Q. Pei and S. Chiba, Mater. Sci. Eng., C, 2000, 11, 89–100 CrossRef.
- H. Böse, D. Uhl and R. Rabindranath, Proc. SPIE, 2012, 8340, 83402E–83411E CrossRef.
- M. Fujino, T. Hisaki, M. Fujiki and N. Matsumoto, Macromolecules, 1992, 25, 1079–1083 CrossRef CAS.
- J. Chojnowski, M. Cypryk, W. Fortuniak, M. Scibiorek and K. Rozga-Wijas, Macromolecules, 2003, 36, 3890–3897 CrossRef CAS.
- M. Cypryk, B. Delczyk, A. Juhari and K. Koynov, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1204–1216 CrossRef CAS.
- L. M. Yi, X. L. Zhan, F. Q. Chen, F. Du and L. B. Huang, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4431–4438 CrossRef CAS.
- A. S. Palsule and Y. Poojari, Polymer, 2010, 51, 6161–6167 CrossRef CAS.
- H. Li, L. Zhang, L. Cheng, Y. Wang, Z. Yu, M. Huang, H. Tu and H. Xia, J. Mater. Sci., 2008, 43, 2806–2811 CrossRef CAS.
- M. Alexandru, M. Cristea, M. Cazacu, A. Ioanid and B. C. Simionescu, Polym. Compos., 2009, 30, 751 CrossRef CAS.
- J. Chojnowski, M. Cypryk, W. Fortuniak, M. Scibiorek and K. Rozga-Wijas, Macromolecules, 2003, 36, 3890–3897 CrossRef CAS.
- A. K. Jonscher, J. Phys. D: Appl. Phys., 1999, 32, R57–R70 CrossRef CAS.
- J. W. Williams, J. Phys. Chem., 1932, 36, 437–443 CrossRef CAS.
- For DMA characterization and cyclic actuation tests of VHB foil, the reader is referred to: D. M. Opris, M. Molberg, C. Walder, Y. S. Ko, B. Fischer and F. A. Nüesch, Adv. Funct. Mater., 2011, 21, 3531–3539 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR and IR spectra, GPC data, DSC curves, self-healing of an actuator. See DOI: 10.1039/c5ra16132k |
| ‡ Present address: Petru Poni Institute of Macromolecular Chemistry, Aleea Grigore Ghica Voda 41A, Iasi, 700487, Romania. |
| This journal is © The Royal Society of Chemistry 2015 |