
5-Phenyl-dipyrromethane and 5-(4-pyridyl)-dipyrromethane as modular building blocks for bio-inspired conductive molecularly imprinted polymer (cMIP). An electrochemical and piezoelectric investigation†
Abstract
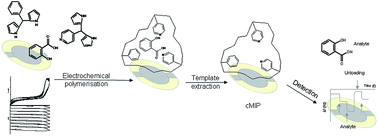
5-Phenyl-dipyrromethane (5-ph-DP) and 5-(4-pyridyl)dipyrromethane (5-py-DP) are proposed, for the first time, as electroactive building blocks for the preparation of sensors based on molecularly imprinted conductive polymers (cMIP). This paper reports the electrochemical and gravimetric investigation on 5-phenyl-dipyrromethane and 5-(4-pyridyl)dipyrromethane and it demonstrates their ability to form both conductive homo-polymers (cMIP) and co-polymers (co-cMIP). The template salicylic acid (SA) was reversibly and selectively incorporated in the obtained synthetic pockets as proved by both voltammetric and piezoelectric investigation. Moreover, the sensitivity of co-cMIP was higher compared to the two homopolymers. The analytical performances confirm that dipyrromethanes, properly functionalized, can be used as electroactive amino acid-like monomers, to prepare bio-inspired imprinted polymers.
 Please wait while we load your content...
Please wait while we load your content...

