
Two-dimensional asynchronous spectrum with auxiliary cross peaks in probing intermolecular interactions†
Xiaopei Liabc,
Anqi Hebd,
Kun Huang*a,
Huizhou Liua,
Ying Zhaoe,
Yongju Weif,
Yizhuang Xu*bd,
Isao Nodabg and
Jinguang Wub
aInstitute of Process Engineering, Chinese Academy of Sciences, 100190, P. R. China. E-mail: khuang@ipe.ac.cn
bBeijing National Laboratory for Molecular Sciences, State Key Laboratory for Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: xyz@pku.edu.cn
cDalian Polytechnic University, Dalian 116034, P. R. China
dNinhai Doubly Advanced Martial Co, Ltd., Ninhai, 315602, China
eInstitute of Chemistry, Chinese Academy of Sciences, 100190, P. R. China
fCollege of Chemistry and Material Science, Hebei Normal University, Shijiazhuang, 050016, P. R. China
gDepartment of Materials Science and Engineering, University of Delaware, Newark, Delaware 19716, USA
First published on 30th September 2015
Abstract
A new approach called “asynchronous spectrum with auxiliary peaks (ASAP)” is proposed for generating a 2D asynchronous spectrum to investigate the intermolecular interaction between two solutes (P and Q) dissolved in the same solution. In the ASAP approach, a virtual substance S with an isolated peak assumed to be at νS is introduced, while the characteristic peaks of P and Q are actually observed at νP and νQ. The concentrations series of P, Q and S are specifically designed so that a spectral portion that has nothing to do with the intermolecular interaction between P and Q is completely removed from the 2D asynchronous spectrum. Auxiliary cross peaks around (νP, νS) and (νQ, νS) can be used to reveal spectral variation caused by intermolecular interaction, which cannot be observed on conventional cross peaks appearing around the spectral coordinates (νP, νP), (νP, νQ), (νQ, νP), (νQ, νQ). For example, variation of the absorptivity of P caused by an intermolecular interaction between P and Q can be probed from the auxiliary cross peaks around (νP, νS) when Q does not even have any characteristic peak in the observed spectral range.
1. Introduction
Intermolecular interaction, which occurs ubiquitously in nature, is of the utmost importance with regard to basic science as well as to applications in organic, biological, medicinal and materials chemistry.1–10 Investigation of the intermolecular interaction has become one of the most active topics in the past two decades.11–22 For example, non-covalent interactions are at the root of the whole field of supra-molecular chemistry that leads to the formation of highly complex and fascinating structures.23 In catalysis, the impact of a weak interaction between ligand and substrate on controlling the reactivity has been recognized.24 In the field of pharmaceuticals, a key step is to rationalize and optimize the interactions between a potential drug and a relevant receptor.25 In protein chemistry, the exploration and comprehension of noncovalent bond interactions are key to predicting the pathways of protein folding and quantifying the relative thermodynamic stability of intermediate and final states.26 In comparison to a plethora of theoretical calculations of the intermolecular interactions, experimental studies on these intriguing intermolecular interactions are still quite limited.Two-dimensional (2D) correlation spectroscopy is a powerful spectroscopic technique proposed by Noda in the late 1980s27–30 and has attracted extensive application in a variety of research fields for the past 25 years.31–47 In 2D correlation spectroscopy, some forms of perturbation are applied to the sample, which imparts variations of spectral signals (called dynamic spectra). Based on cross-correlation analysis, the dynamic spectra are then transformed into a spectrum with two independent spectral variable axes (2D correlation spectrum). In general, 2D correlation spectra are classified into two types: synchronous correlation spectra and asynchronous correlation spectra obtained by using different cross-correlation methods. Owing to the enhancement of the spectral resolution by spreading the peaks over the second dimension, subtle changes of the sample, not readily seen in the original data set, can be visualized in terms of cross peaks in the 2D correlation spectra.
One of the most important features of 2D correlation spectra is that cross peaks in 2D correlation spectra can potentially be used to characterize intermolecular interactions.27,28 However, this approach suffers from the following problem: interfering cross peaks due to other sources of correlation may also arise even if there are no intermolecular interactions. This makes the mere appearance of cross peaks in 2D correlation spectra difficult to use as a reliable tool to characterize intermolecular interactions.
To address the problem, we proposed an orthogonal sample design scheme (OSD) approach in our previous work.48–53 The brief description of the OSD approach by Noda in his recent review43 is given as follows: the basic concept of OSD is to use a well-designed set of concentration series for two different constituents in solution mixtures, such that patterns of concentration variations of the two species will become mathematically orthogonal to each other. The imposed orthogonality will break down when the apparent deviation from the Beers–Lambert law, often associated with the presence of specific intermolecular interactions, is observed. Thus, the OSD technique becomes a very sensitive probe for the possible presence of specific molecular interactions. It should also be pointed out that OSD may be viewed as a form of multiple perturbation 2D correlation method, since two separate concentration variations are simultaneously imposed as perturbations. The unique feature of OSD is to proactively design the selective perturbation conditions to maximize the information content of the resulting 2D correlation spectra.
Following this original idea, we have introduced asynchronous orthogonal sample design (AOSD),54–56 double orthogonal sample design (DOSD)57 and double asynchronous orthogonal sample design (DAOSD)58–66 schemes to further enhance the ability of 2D correlation spectroscopy to reveal spectral variations of the characteristic peaks of solutes caused by intermolecular interactions.
The chemical systems applicable for the OSD and related approaches are solutions containing two solutes (P and Q). The spectral coordinate of a characteristic peak from P is given by νP, and that from Q is νQ. Cross-peaks around (νP, νQ) in the 2D correlation spectrum are used to reflect intermolecular interactions between P and Q. That is to say, both P and Q possessing characteristic peaks is the prerequisite to apply OSD and relevant techniques to probe the intermolecular interactions between P and Q. In many cases, however, only one solute possesses a characteristic peak, while another solute does not have any characteristic peak within the observed spectral region. Consequently, the OSD and related approaches cannot be used directly to probe intermolecular interactions between two solutes in these chemical systems.
In our recent paper,67 we developed a new approach to probe intermolecular interactions between P and Q dissolved in the same solutions. In the system, only P possesses a characteristic peak at spectral coordinate νP, while Q does not possess any characteristic peak. Based on mathematical analysis, computer simulation and experiments on a real chemical system, we demonstrated that cross peaks around the spectral coordinate (νP, νP) in asynchronous correlation spectra can also be used to reflect intermolecular interactions between P and Q. Moreover, the patterns of cross peaks around the coordinate (νP, νP) can be used to reveal subtle variations of peak position and bandwidth of the characteristic peak of P caused by intermolecular interactions. Unfortunately, variations on absorptivity of the characteristic peak of P cannot be reflected by the pattern of cross peaks around the coordinate (νP, νP).
The fact that the patterns of cross peaks fail to reflect variation of absorptivity brings about the following two problems: (1) the failure prevents us from obtaining comprehensive information on the spectroscopic behavior of the characteristic peak of P under intermolecular interactions. (2) The inability to reflect the variation of absorptivity means the approach is at risk of making incorrect conclusions concerning whether intermolecular interactions occur or not in some special cases. If intermolecular interactions only bring about changes on the absorptivity of the characteristic peak of P, no cross peaks can be observed around the coordinate (νP, νP) in the asynchronous correlation spectrum.
In order to solve this problem, a new method, called the “asynchronous spectrum with auxiliary peaks” (ASAP) approach, is proposed. In the ASAP approach, a virtual substance (denoted as S) with an isolated characteristic peak at coordinate νS is introduced. The cross peaks around (νS, νP) and (νS, νQ) are called auxiliary cross peaks. Mathematical analysis, computer simulation and experiment on a real chemical system were carried out. The results demonstrate that variations on absorptivity of the characteristic peak of P can be reflected by the auxiliary cross peaks around (νS, νP) when only P possesses a characteristic peak.
2. Experimental
2.1 Description of the model system used in the ASAP approach
The chemical system considered here consists of n solutions containing two solutes (P and Q). Variable concentrations are used as an external perturbation to construct 2D asynchronous correlation spectra. In addition, a virtual solute denoted as S is also introduced in association with each solution. The initial concentrations of P, Q and S are denoted as:
 | (1a) |
 | (1b) |
 | (1c) |
When there are intermolecular interactions between P and Q, part of P undergoes subtle structural variation and converts to U and part of Q converts to V. This inter-conversion can be expressed by the following equilibrium where K is the equilibrium constant. The so-called solute S is a virtual substance, and it does not interact with either P or Q.
 | (2) |
For the ith solution, the corresponding spectrum is given by eqn (3).
| Ai(ν) = fP(ν)Ci(eq)P + fQ(ν)Ci(eq)Q + fU(ν)Ci(eq)U + fV(ν)Ci(eq)V + fS(ν)Ci(eq)S | (3) |
For each of P, Q, U, V and S, the spectral function is a single peak function that is represented by a Gaussian function as shown in eqn (4).
 | (4) |
Since S does not interact with other solutes, we have:
| Ci(init)S = Ci(eq)S | (5) |
Based on eqn (2), the following two expressions can be obtained.
| Ci(eq)P = Ci(init)P − Ci(eq)U | (6a) |
| Ci(eq)Q = Ci(init)Q − Ci(eq)V | (6b) |
Thus, eqn (3) also can be expressed as:
| Ai(ν) = fP(ν)Ci(init)P + [fU(ν) − fP(ν)]Ci(eq)U + fQ(ν)Ci(init)Q + [fV(ν) − fQ(ν)]Ci(eq)V + fS(ν)Ci(init)S | (7) |
After removing the average value over all solution samples at each wavelength, the dynamic spectrum of the ith solution can be expressed as eqn (8).
Ãi(ν) = fP(ν)![[C with combining tilde]](https://www.rsc.org/images/entities/i_char_0043_0303.gif) i(init)P + [fU(ν) − fP(ν)]i(eq)U + fQ(ν)i(init)Q + [fV(ν) − fQ(ν)]i(eq)V + fS(ν)i(init)S i(init)P + [fU(ν) − fP(ν)]i(eq)U + fQ(ν)i(init)Q + [fV(ν) − fQ(ν)]i(eq)V + fS(ν)i(init)S
| (8) |
|
i(init)P = Ci(init)P − Cinit(av)P
| (9a) |
|
i(init)Q = Ci(init)Q − Cinit(av)Q
| (9b) |
|
i(eq)U = Ci(eq)U − Ceq(av)U
| (9c) |
|
i(eq)V = Ci(eq)V − Ceq(av)V
| (9d) |
|
i(init)S = Ci(init)S − Cinit(av)S
| (9e) |
i(init)P, i(init)Q and i(init)S are the dynamic initial concentrations of P, Q and S in the ith solution. i(eq)U and i(eq)V are the dynamic equilibrium concentrations of U and V in the ith solution.
 | (10a) |
 | (10b) |
 | (10c) |
 | (10d) |
 | (10e) |
Asynchronous correlation spectra can be constructed based on eqn (8) and (11)
 | (11) |
 and
and  are the dynamic spectral vector
are the dynamic spectral vector  at the spectral coordination ν1 and ν2, respectively.
at the spectral coordination ν1 and ν2, respectively.
In the computer simulation on the model system, the simulated 1D spectra were generated via a program written in our lab with the MATLAB software. All asynchronous correlation spectra were calculated based on the algorithm by Noda30 using the software of MATLAB.
2.2 Experiment on a real chemical system
3. Results and discussion
Scheme 1 illustrates the 1D spectra used in constructing the asynchronous correlation spectrum. Since S is a virtual substance, the characteristic peak of S appears as gray lines. In principle, the molar absorptivity, peak position and bandwidth of the characteristic peak of S can be arbitrary. In the experiment, the peak of S does not overlap the characteristic peaks of P and Q. | ||
| Scheme 1 Schematic diagram of the simulated 1D spectra used in constructing an asynchronous correlation spectrum. S is a virtual substance, and its characteristic peak appears as gray lines. It should be pointed out that the spectral region between 200 cm−1 and 0 cm−1 in this scheme is a virtual frequency region. | ||
The cross peaks in the asynchronous correlation spectrum based on the ASAP approach can be divided into three spectral domains, as shown in Scheme 2. Domain I contains the cross peaks around the spectral coordinates (νP, νP), (νQ, νQ), (νP, νQ) and (νQ, νP). These cross peaks are conventional cross peaks in an asynchronous correlation spectrum. Cross peaks located around (νS, νP) and (νS, νQ) in domain II and cross peaks around (νP, νS) and (νQ, νS) in domain III are auxiliary cross peaks. According to the basic properties of asynchronous correlation spectra, the auxiliary cross peaks in domain III are anti-symmetric to those in domain II with respect to the diagonal. Thus, we focus on the auxiliary cross peaks in domain II in the following part.
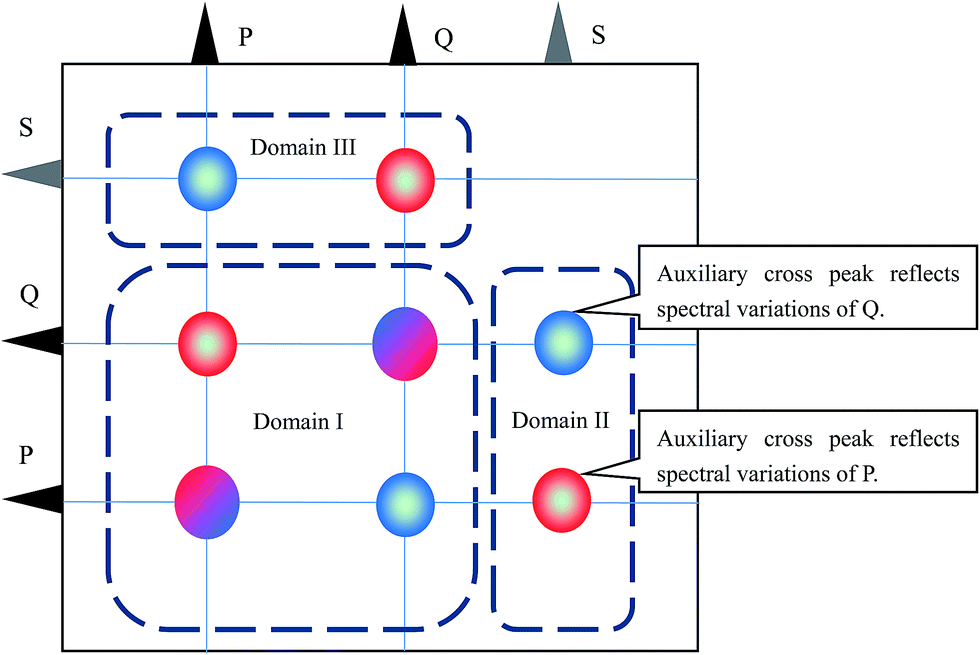 | ||
| Scheme 2 Asynchronous correlation spectrum generated by using the ASAP approach. Cross peaks can be classified into three domains. Domain I contains the conventional cross peaks in an asynchronous correlation spectrum. Both domain II and domain III contain auxiliary cross peaks. Since cross peaks in domain III are anti-symmetric to the cross peaks in domain II with respect to diagonal, only auxiliary cross peaks in domain II are discussed. | ||
3.1 Basic properties of the ASAP approach
It is assumed that S does not interact with P or Q since it is a virtual substance. We try to use the auxiliary cross peaks around (νS, νP) and (νS, νQ) to reflect the intermolecular interactions between P and Q. In an ideal asynchronous correlation spectrum generated by using the ASAP approach, the auxiliary cross peaks should possess the following two properties: (I): no auxiliary cross peak can be produced around (νS, νP) and (νS, νQ) when there are no intermolecular interactions between P and Q. (II): when intermolecular interactions occur between P and Q, auxiliary cross peaks around (νS, νP) or (νS, νQ) should be present. Moreover, the variations of spectral function caused by the conversion from P to U and from Q to V can be manifested by the auxiliary cross peaks around (νS, νP) or (νS, νQ).To achieve the above goals, mathematical analysis on the auxiliary cross peaks around (νS, νP) or (νS, νQ) is carried out. Herein, ν1 is in the spectral region of the characteristic peak of S, while ν2 is in the spectral region of the characteristic peaks of P, Q, U and V. Ψ(ν1, ν2) is calculated by combining eqn (8) and (11) and its expression can be expressed as eqn (12). The expressions of the twenty-five terms in eqn (12) are given in the appendix.
 | (12) |
Since the characteristic peak of S is intentionally set not to overlap with characteristic peaks of P, Q, U, V, we have:
 | (13) |
Based on eqn (13), the values of R1 ∼ R20 and R25 in eqn (12) are all zero, and thus only four terms R21 ∼ R24 are left.
When there are no intermolecular interactions between P and Q, the equilibrium concentrations of U and V, which are the products of intermolecular interactions between P and Q, should be zero. Thus we have:
 | (14) |
According to eqn (14), the values of R23 and R24 are zero when there are no intermolecular interactions between P and Q.
 | (15) |
The corresponding Ψ(ν1, ν2) changes into eqn (16).
 | (16) |
Since no intermolecular interactions occur between P and Q, Ψ(ν1, ν2) should be zero. To make Ψ(ν1, ν2) be zero, a feasible way is to make both R21(ν1,ν2) and R22(ν1,ν2) be zero. This can be achieved by setting the initial concentrations of S to be linearly proportional to the initial concentrations of P and Q simultaneously (eqn (17a) and (17b)). Mathematical analysis to support this statement is given in detail in the appendix.
| aCi(init)S + bCi(init)Q = c | (17a) |
| mCi(init)S + hCi(init)P = d | (17b) |
Thus, property I is achieved if the concentration series of P, Q and S satisfy eqn (17a) and (17b). That is to say, no auxiliary cross peaks are produced around (νS, νP) and (νS, νQ) when there are no intermolecular interactions between P and Q.
By selecting the concentration series P, Q and S based on eqn (17a) and (17b), the auxiliary cross peaks can be expressed as eqn (18) for the chemical system where intermolecular interactions occur between P and Q.
 | (18) |
According to eqn (4) and (18) changes into eqn (19)
 | (19) |
Eqn (19) can be expressed as a summation of four parts. The first term contains gU(ν1) − gP(ν1). That is to say, it reflects variations of bandwidth and peak position of P. The second term contains (εV − εQ), demonstrating that it is relevant to the variations of absorptivity of P. Similarly, the third term reflects the variations of bandwidth and peak position of Q and the fourth term is relevant to the variation of absorptivity of Q. Thus, the auxiliary cross peaks in the ASAP approach do reflect the variation of spectral function of P and Q caused by intermolecular interaction. Therefore, property II of the auxiliary cross peak is also achieved.
3.2 The application of the ASAP approach in reflecting the variation of absorptivity when only one substance involving intermolecular interactions possesses characteristic peaks
Equipped with the ASAP approach, we try to establish a method to reveal the variation of absorptivity of P when Q does not possess any characteristic peak in the spectral region.Considering a chemical system where P possesses a characteristic peak at νP but Q has no characteristic peak, we have proved that cross peaks around the coordinate (νP, νP) near the main diagonal in an asynchronous correlation spectrum can be used to characterize the intermolecular interactions between P and Q. In our previous work,67 we demonstrated that the cross peaks around the coordinate (νP, νP) can be expressed as eqn (20).
 | (20) |
Based on the mathematical property of the Hilbert–Noda transformation, matrix N in eqn (21), H1(x, y) and H2(x, y) can be combined into one term.
 | (21) |
![[A with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/b_i_char_0041_20d1.gif) and
and ![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/b_i_char_0042_20d1.gif) can be arbitrary n-dimensional vectors.
can be arbitrary n-dimensional vectors.
Eqn (20) can be expressed as eqn (22).
 | (22) |
As shown in eqn (22), the pattern of cross peaks around (νP1, νP2) can reflect the variations of g(ν) that are relevant to peak position and bandwidth. Although the variation of absorptivity is related to the intensity of cross peak, it is hard to retrieve information on the variations of absorptivity from the intensity of the cross peak, since the intensity of a cross peak is affected by a variety of factors. These factors are difficult to measure accurately.
To solve the problem, the ASAP approach is adopted, and we try to obtain the variation of the absorptivity of the characteristic peak of P from the auxiliary cross peak. Since both Q and V do not show any characteristic peak, we have fQ(ν1) = 0 and fV(ν1) = 0. Thus, the R24 term in eqn (18) is zero. Consequently, the auxiliary cross peaks can be expressed as eqn (23).
 | (23) |
As shown in eqn (23), the auxiliary cross peak is composed of two parts. We notice that the second part contains (εU − εP) term, which reflects the variation of absorptivity of the characteristic peak of P under intermolecular interactions. Therefore, the problem that a variation of the absorptivity of the characteristic peak of P cannot be reflected when Q does not possess any characteristic peak is addressed by using the ASAP approach. Information on the variation of absorptivity can be retrieved from the pattern of auxiliary cross peaks around (νS, νP).
First, we carry out a computer simulation on a model chemical system to show how variation of the absorptivity can be obtained by using the ASAP approach. In this simulated system, intermolecular interactions between P and Q only cause the variation of absorptivity of P, and the corresponding K value is arbitrarily set as 0.01 here. The spectral parameters of P, U and S are given in Table 1. The concentrations of P, Q and S are listed in Table 2.
| Number | CP | CQ | CS |
|---|---|---|---|
| 1 | 2.00 | 5.00 | 10.00 |
| 2 | 0.00 | 3.00 | 12.00 |
| 3 | 4.00 | 7.00 | 8.00 |
| 4 | 7.00 | 10.00 | 5.00 |
As shown in Fig. 1, no cross peak in domain I is observed, indicating that a variation of absorptivity of the characteristic peak of P cannot be reflected by the conventional cross peaks in an asynchronous correlation spectrum. Furthermore, this result demonstrates that conventional cross peaks fail to detect intermolecular interaction between P and Q. However, a single auxiliary cross peak can be clearly observed around (100, 350) in domain II of Fig. 1. This result demonstrates that intermolecular interactions between P and Q can be manifested by the presence of an auxiliary cross peak. Moreover, the pattern of auxiliary cross peaks is also helpful to reveal the variation of absorptivity. As shown in domain I of Fig. 1, no cross peak around (350, 350) is observable, indicating that intermolecular interactions do not produce variations in either peak position or bandwidth of the characteristic peak of P. Thus, the first term in eqn (23) is zero and the second term relevant to the variation of absorptivity is left. According to eqn (23), the second term will produce a single auxiliary cross peak. As shown in domain II of Fig. 1, a single auxiliary cross peak is observed, confirming that the variation of absorptivity is revealed.
 | ||
| Fig. 1 Asynchronous correlation spectrum based on the ASAP approach when just P possesses a characteristic peak and intermolecular interactions induce a variation in the absorptivity of P. It should be pointed out that the spectral region between 200 cm−1 and 0 cm−1 in this figure is a virtual frequency region. | ||
Then we apply the ASAP approach on a real chemical system. In the real chemical system, coordination between Li+ and benzo-15-crown-5 is probed. First, a series of methanol solutions containing lithium chloride (denoted as LC) and benzo-15-crown-5 (denoted as BC) was prepared. The concentrations of benzo-15-crown-5 and lithium chloride are listed in Table 3. The concentrations of S in the four solutions listed in Table 3 are 0.00, 0.08, 0.19 and 0.30 mol L−1 respectively. The peak position, bandwidth and absorptivity of the characteristic peak of the virtual substance (S) are set to be 1450.00, 10.00 and 1.00 respectively. FT-IR spectra of the solutions were recorded and are shown in Fig. 2A. The band located at around 1597 cm−1 is assigned to vibration of the skeleton of the aromatic ring and used as a characteristic peak of benzo-15-crown-5. In the FTIR spectra, spectral data below 1500 cm−1 are truncated and spectra of a virtual substance S are put in the spectral region between 1500 and 1400 cm−1. The peaks shown in gray refer to the virtual substance S. Li+ does not exhibit any absorption peak in the FTIR spectrum. The asynchronous correlation spectrum based on the ASAP approach is constructed by using the 1D spectra of Fig. 2A and is shown in Fig. 2B. A pair of cross peaks in domain I can be clearly observed in Fig. 2B.
| Number | CBC (mol L−1) | CLC (mol L−1) |
|---|---|---|
| 1 | 0.30 | 0.00 |
| 2 | 0.22 | 0.08 |
| 3 | 0.11 | 0.19 |
| 4 | 0.00 | 0.30 |
 | ||
| Fig. 2 (A) FT-IR spectra of the benzo-15-crown-5 of the solutions; (B) 2D asynchronous correlation spectrum generated by using the 1D spectra in (A) based on the ASAP approach. The spectral region between 1500 cm−1 and 1400 cm−1 is a virtual frequency region. In (B), the absolute intensity of the positive peak is larger than that of the negative peak (this is manifested by the fact that the number of contours in the positive auxiliary peak is larger than that in the negative auxiliary peak). | ||
According to our previous work,48–67 the following experiment is performed: benzo-15-crown-5 is dissolved in methanol alone. A good linear relationship between the absorbance of the 1597 cm−1 band and the concentration of benzo-15-crown-5 can be obtained when the concentration range of benzo-15-crown-5 is between 0.00 mol L−1 and 0.30 mol L−1 (ESI†). Since the concentrations of benzo-15-crown-5, listed in Table 3, are within the above concentration range, the possibility that the conventional cross peaks in domain I of Fig. 2B are caused by the interaction between benzo-15-crown-5 and methanol can be safely precluded. That is to say, it is the coordination between Li+ and benzo-15-crown-5 that brings about the structural variation on the aromatic ring and produces the cross peaks in the corresponding asynchronous correlation spectrum. This spectral pattern suggests that coordination between Li+ and benzo-15-crown-5 brings about a blue shift of the 1597 cm−1 band. The result obtained from the conventional cross peak in domain I is consistent with that shown in Fig. 2A.
However, whether the absorptivity of the 1597 cm−1 band varies or not remains unknown. Herein the auxiliary cross peak around (1450, 1597 cm−1) in domain II is used to check whether the intermolecular interactions between benzo-15-crown-5 and Li+ cause variations of absorptivity other than peak position. According to the 1D spectra in Fig. 2A and the conventional cross peak in domain I of Fig. 2B, we learn that the interactions between benzo-15-crown-5 and Li+ make the characteristic peak of benzo-15-crown-5 undergo a blue-shift (ΔxU > 0). In domain II, a pair of vertical auxiliary cross peaks can be observed. One auxiliary cross peak is positive and the other is negative. The spectral pattern of the auxiliary cross peak also indicates that the 1597 cm−1 band undergoes a band-shift. When we examine the auxiliary cross peaks carefully, it is noticed that the absolute intensity of the positive auxiliary cross peak is slightly larger than that of the negative auxiliary cross peak. The pattern of cross peaks around the coordinate (1597, 1597 cm−1) demonstrates that coordination between the lithium ion and benzo-15-crown-5 cannot produce an observable variation in the bandwidth of the 1597 cm−1 peak. If coordination only brings about band-shift of the 1597 cm−1 peak, the absolute intensities of the pair of the auxiliary cross peaks should be the same. This is not the case in Fig. 2B, suggesting that the absorptivity of the 1597 cm−1 peak also changes as coordination occurs between benzo-15-crown-5 and lithium ion.
Thus, we performed a computer simulation on three model systems to mimic the spectral behavior of the benzo-15-crown-5/lithium chloride system. The peak parameters of P, U and S in the three model systems are listed in Table 4. The corresponding 2D asynchronous spectra are shown in Fig. 3. It is found that the pattern of auxiliary cross peaks in Fig. 2B is quite similar to that shown in Fig. 3C. This result demonstrates that the absorptivity of the 1597 cm−1 band also increases upon coordinating with lithium ion. That is to say, coordination between Li+ and benze-15-crown-5 not only makes the 1597 cm−1 band undergo a blue shift but also brings about a slight increment of its absorptivity.
| Model system I | Model system II | Model system III | |
|---|---|---|---|
| XP (cm−1) | 1597.00 | 1597.00 | 1597.00 |
| WP (cm−1) | 10.00 | 10.00 | 10.00 |
| εP | 1.00 | 1.00 | 1.00 |
| XU (cm−1) | 1600.00 | 1600.00 | 1600.00 |
| WU (cm−1) | 10.00 | 10.00 | 10.00 |
| εU | 0.97 | 1.00 | 1.03 |
| XS (cm−1) | 1450.00 | 1450.00 | 1450.00 |
| WS (cm−1) | 10.00 | 10.00 | 10.00 |
| εS | 1.00 | 1.00 | 1.00 |
| XU − XP (cm−1) | 3.00 | 3.00 | 3.00 |
| WU − WP (cm−1) | 0.00 | 0.00 | 0.00 |
| εU − εP | −0.03 | 0.00 | 0.03 |
 | ||
| Fig. 3 2D asynchronous correlation spectra generated by using the ASAP approach on three model systems to simulate the spectral variation of the lithium chloride/benzo-15-crown-5 system. The spectral region between 1500 cm−1 and 1400 cm−1 in figure is a virtual frequency region. Red contours mean the cross peaks are positive and blue contours mean the cross peaks are negative. The pattern of the auxiliary cross peak around (1450, 1597 cm−1) in (C) is similar to that shown in Fig. 2B. The absolute intensity of the positive peak is larger than that of the negative peak (this is manifested by the fact that the number of contours in the positive auxiliary peak is larger than that in the auxiliary negative peak). | ||
In summary, we propose the ASAP approach, where a virtual substance(S) is introduced into solutions containing two solutes (P and Q), is a useful technique. Under a suitable concentration series, auxiliary cross peaks around (νS, νP) and (νS, νQ) can be used to reflect intermolecular interactions between P and Q. By using the ASAP approach, variations of absorptivity of the characteristic peak of P can be retrieved when Q has no characteristic peak in the spectral region.
Acknowledgements
This project is financially supported by the National Natural Science Foundation of China (No. 51373003, 51074150, 51574213), Natural Science Foundation of Hebei Province (No. B2014205129) and the Key Project of Chinese National Programs for Fundamental Research and Development (973 Programs No. 2012CBA01203, 2013CB632602).References
- J. Yu, L. S. Bi, R. K. Kalia and P. Vashishta, Intermolecular and intramolecular phonons in solid C60-effects of orientational disorder and pressure, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 5008–5019 CrossRef CAS.
- S. Tsuzuki and T. Uchimaru, Magnitude and physical origin of intermolecular interactions of aromatic molecules: recent progress of computational studies, Curr. Org. Chem., 2006, 10, 745–762 CrossRef CAS.
- I. Nudelman, S. R. Akabayov, E. Schnur, Z. Biron, R. Levy, Y. Xu, D. Yang and J. Anglister, Intermolecular interactions in a 44 kDa interferon–receptor complex detected by asymmetric reverse-protonation and two-dimensional NOESY, Biochemistry, 2010, 49, 5117–5133 CrossRef CAS PubMed.
- N. Veljkovic, S. Glisic, V. Perovic and V. Veljkovic, The role of long-range intermolecular interactions in discovery of new drugs, Expert Opin. Drug Discovery, 2011, 6, 1263–1270 CrossRef CAS PubMed.
- Y. F. Xie, Y. Y. Huang, W. Z. Wang, G. Q. Liu and R. Zhao, Dynamic interaction between melamine and cyanuric acid in artificial urine investigated by quartz crystal microbalance, Analyst, 2011, 136, 2482–2488 RSC.
- M. Maniruzzaman, D. J. Morgan, A. P. Mendham, J. Pang, M. J. Snowden and D. Douroumis, Drug-polymer intermolecular interactions in hot-melt extruded solid dispersions, Int. J. Pharm., 2013, 443, 199–208 CrossRef CAS PubMed.
- K. Ruanjaikaen and A. L. Zydney, Intermolecular interactions during ultrafiltration of pegylated proteins, Biotechnol. Prog., 2013, 29, 655–663 CrossRef CAS PubMed.
- H. Wang, N. Chen and Q. Wang, Interaction between β-tricalcium phosphate and poly(vinyl alcohol) and its effect on the thermal and mechanical properties of poly(vinyl alcohol), Chem. J. Chin. Univ., 2014, 35, 1810–1815 CAS.
- X. F. Zhang, L. Chen, Q. F. Yang, X. R. Sun, H. B. Chen, G. Yang and Y. L. Tang, Investigation on the cyanine dyes supramolecular assembly and chiral inducement by fulvic acid, Spectrosc. Spectral Anal., 2014, 34, 3045–3050 CAS.
- F. Sakai, Z. W. Ji, J. H. Liu, G. S. Chen and M. Jiang, A novel supramolecular graft copolymer via cucurbit 8 uril-based complexation and its self-assembly, Chin. Chem. Lett., 2013, 24, 568–572 CrossRef CAS PubMed.
- N. Fatkullin, A. Gubaidullin, C. Mattea and S. Stapf, On the theory of the proton free induction decay and Hahn echo in polymer systems: the role of intermolecular magnetic dipole–dipole interactions and the modified Anderson-Weiss approximation, J. Chem. Phys., 2012, 137, 224907 CrossRef CAS PubMed.
- J. A. Molina-Bolivar, F. Galisteo-Gonzalez, C. Carnero Ruiz, M. Medina-O'Donnell and A. Parra, Spectroscopic investigation on the interaction of maslinic acid with bovine serum albumin, J. Lumin., 2014, 156, 141–149 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Interactions with aromatic rings in chemical and biological recognition, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- M. Aoudia, T. Al-Maamari and F. Al-Salmi, Intramolecular and intermolecular ion–dipole interactions in sodium lauryl ether sulfates (SLES) self-aggregation and mixed micellization with Triton X-100, Colloids Surf., A, 2009, 335, 55–61 CrossRef CAS PubMed.
- J. S. Li, An ab initio study of intermolecular interaction of hydrogen fluoride tetramer, J. Theor. Comput. Chem., 2006, 5, 187–196 CrossRef CAS.
- J. Liu, S. X. Liu, Y. L. Gao, X. K. Fan, Y. Guan, S. F. Weng, Z. L. Yang, Y. Z. Xu and J. G. Wu, On the interaction between PVP and europium benzenesulfonate, Spectrosc. Spectral Anal., 2013, 33, 1487–1490 CAS.
- S. R. Wu, B. Jin, N. Zhang, Y. Liu, X. J. Liu, S. Q. Li and L. H. Shanggan, New monomethine cyanine dye and its interaction with different DNA forms, Chem. J. Chin. Univ., 2014, 35, 2085–2092 CAS.
- T. Madrakian and S. Heidari, Interaction of benzene-1,3-disulfonylamid-kriptofix 22 with iodine in chloroform and dichloromethane solutions, Chin. Chem. Lett., 2014, 25, 1375–1378 CrossRef CAS PubMed.
- J. Sun, J. S. Yu, S. Jin, X. Zha, Y. Q. Wu and Z. W. Yu, Interaction of synthetic HPV-16 capsid peptides with heparin: thermodynamic parameters and binding mechanism, J. Phys. Chem. B, 2010, 114, 9854–9861 CrossRef CAS PubMed.
- Q. Z. Li, N. N. Wang and Z. W. Yu, Solvent effect on the role of methyl groups in formation of O⋯HO hydrogen bond in dimethyl ether–methanol complex, J. Mol. Struct.: THEOCHEM, 2008, 862, 74–79 CrossRef CAS PubMed.
- R. Wang, Q. Z. Li, R. G. Wu, G. S. Wu and Z. W. Yu, Molecular interactions between pyrazine and n-propanol, chloroform, or tetrahydrofuran, Spectrochim. Acta, Part A, 2008, 70, 793–798 CrossRef PubMed.
- C. Schmuck and J. Lex, C2H⋯O Interactions as Isofunctional Replacements for N2H⋯O Interactions 2 Dimer Formation of Methyl 5-Amidopyrrole-2-Carboxylates in the Solid State, Eur. J. Org. Chem., 2001, 1519–1523 CrossRef CAS.
- M. Roman, C. Cannizzo, T. Pinault, B. Isare, B. Andrioletti, P. van der Schoot and L. Bouteiller, Supramolecular Balance: Using Cooperativity to Amplify Weak Interactions, J. Am. Chem. Soc., 2010, 132, 16818–16824 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Fluorine in Pharmaceuticals: Looking Beyond Intuition, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- E. Shakhnovich, Protein Folding Thermodynamics and Dynamics: Where Physics, Chemistry, and Biology Meet, Chem. Rev., 2006, 106, 1559–1588 CrossRef CAS PubMed.
- I. Noda, Two-dimensional infrared-spectroscopy, J. Am. Chem. Soc., 1989, 111, 8116–8118 CrossRef CAS.
- I. Noda, 2-Dimensional infrared (2D IR) spectroscopy – theory and applications, Appl. Spectrosc., 1990, 44, 550–561 CrossRef CAS.
- I. Noda, Generalized 2-dimensional correlation method applicable to infrared, Raman, and other types of spectroscopy, Appl. Spectrosc., 1993, 47, 1329–1336 CrossRef CAS.
- I. Noda, Determination of Two-Dimensional Correlation Spectra Using the Hilbert Transform, Appl. Spectrosc., 2000, 54, 994–999 CrossRef CAS.
- D. L. Elmore and R. A. Dluhy, Beta nu-correlation analysis: a modified two-dimensional infrared correlation method for determining relative rates of intensity change, J. Phys. Chem. B, 2001, 105, 11377–11386 CrossRef CAS.
- S. Shanmukh and R. A. Dluhy, kv Correlation analysis. A quantitative two-dimensional IR correlation method for analysis of rate processes with exponential functions, J. Phys. Chem. A, 2004, 108, 5625–5634 CrossRef CAS.
- R. Dluhy, S. Shanmukh and S. I. Morita, The application of two-dimensional correlation spectroscopy to surface and interfacial analysis, Surf. Interface Anal., 2006, 38, 1481–1496 CrossRef CAS PubMed.
- S. I. Morita, S. Shanmukh, Y. Ozaki and R. A. Dluhy, A general model-based approach to two-dimensional infrared correlation spectroscopy incorporating the global phase angle, Appl. Spectrosc., 2006, 60, 1279–1284 CrossRef CAS.
- D. Pivonka, Vibrational analysis of structure activity relationships (SAR) in molecular binding, Appl. Spectrosc., 2004, 58, 323–331 CrossRef CAS PubMed.
- S. Morita, H. Shinzawa, I. Noda and Y. Ozaki, Perturbation-correlation moving-window two-dimensional correlation spectroscopy, Appl. Spectrosc., 2006, 60, 398–406 CrossRef CAS PubMed.
- H. Shinzawa, K. Awa, I. Noda and Y. Ozaki, Multiple-perturbation two-dimensional near-infrared correlation study of time-dependent water absorption behavior of cellulose affected by pressure, Appl. Spectrosc., 2013, 67, 163–170 CrossRef CAS PubMed.
- H. Shinzawa and I. Noda, Two-dimensional infrared (2D IR) correlation spectroscopy study of self-assembly of oleic acid (OA) in conjunction with partial attenuation of dominant factor by eigenvalue manipulating transformation (EMT), Vib. Spectrosc., 2012, 60, 180–184 CrossRef CAS PubMed.
- H. Park, H. S. Kim and Y. M. Jung, Interionic interactions of binary gels consisting of pyrrolidinium-based zwitterionic compounds and lithium salts, J. Phys. Chem. B, 2011, 115, 1743–1750 CrossRef CAS PubMed.
- H. J. Kim, S. B. Kim, J. K. Kim and Y. M. Jung, Two-dimensional heterospectral correlation analysis of wide-angle X-ray scattering and infrared spectroscopy for specific chemical interactions in weakly interacting block copolymers, J. Phys. Chem. B, 2006, 110, 23123–23129 CrossRef CAS PubMed.
- H. Lee, Y. M. Jung, K. I. Lee, H. S. Kim and H. S. Park, Understanding the unique interaction of amine-containing ionic compounds with SO2 for high absorption capacity, RSC Adv., 2013, 3, 25944–25949 RSC.
- Z. W. Yu, L. Chen, S. Q. Sun and I. Noda, Determination of selective molecular interactions using two-dimensional correlation FT-IR spectroscopy, J. Phys. Chem. A, 2002, 106, 6683–6687 CrossRef CAS.
- I. Noda, Frontiers of two-dimensional correlation spectroscopy. Part 1. New concepts and noteworthy developments, J. Mol. Struct., 2014, 1069, 3–22 CrossRef CAS PubMed.
- I. Noda, Frontiers of two-dimensional correlation spectroscopy. Part 2. Perturbation methods, fields of applications, and types of analytical probes, J. Mol. Struct., 2014, 1069, 23–49 CrossRef CAS PubMed.
- C. Li, J. W. Liu, J. J. Li, F. Shen, Q. S. Huang and H. X. Xu, Studies of 4,4′-diphenylmethane diisocyanate (MDI)/1,4-butanediol (BDO) based TPUs by in situ and moving-window two-dimensional correlation infrared spectroscopy: understanding of multiple DSC endotherms from intermolecular interactions and motions level, Polymer, 2012, 53, 5423–5435 CrossRef CAS PubMed.
- A. Awichi, E. M. Tee, G. Srikanthan and W. Zhao, Identification of Overlapped Near-Infrared Bands of Glucose Anomers Using Two-Dimensional Near-Infrared and Middle-Infrared Correlation Spectroscopy, Appl. Spectrosc., 2002, 56, 897 CrossRef CAS.
- E. M. Tee, A. Awichi and W. Zhao, Probing Microstructure of Acetonitrile–Water Mixtures by Using Two-Dimensional Infrared Correlation Spectroscopy, J. Phys. Chem. A, 2002, 106, 6714–6719 CrossRef CAS.
- J. Qi, H. Z. Li, K. Huang, S. X. Liu, L. M. Yang, Y. Zhao, C. F. Zhang, W. H. Li, J. G. Wu, D. F. Xu, Y. Z. Xu and I. Noda, Orthogonal sample design scheme for two-dimensional synchronous spectroscopy and its application in probing intermolecular interactions, Appl. Spectrosc., 2007, 61, 1359–1365 CrossRef CAS PubMed.
- J. Qi, K. Huang, X. X. Gao, H. Z. Li, S. X. Liu, Y. Zhao, Y. Z. Xu, J. G. Wu and I. Noda, Orthogonal sample design scheme for two-dimensional synchronous spectroscopy: application in probing lanthanide ions interactions with organic ligands in solution mixtures, J. Mol. Struct., 2008, 883, 116–123 CrossRef PubMed.
- H. Z. Li, D. L. Tao, J. Qi, J. G. Wu, Y. Z. Xu and I. Noda, Dipole–dipole interactions in solution mixtures probed by two-dimensional synchronous spectroscopy based on orthogonal sample design scheme, Spectrochim. Acta, Part A, 2014, 124, 697–702 CrossRef CAS PubMed.
- Y. H. Liu, C. F. Zhang, S. X. Liu, Y. Zhao, D. J. Wang, J. G. Wu, Y. Z. Xu and I. Noda, Modified orthogonal sample design scheme to probe intermolecular interactions, J. Mol. Struct., 2008, 883, 124–128 Search PubMed.
- J. Chen, C. F. Zhang, H. Z. Li, Y. F. Liu, W. H. Li, Y. Z. Xu, J. G. Wu and I. Noda, Patterns of cross peaks in 2D synchronous spectrum generated by using orthogonal sample design scheme, J. Mol. Struct., 2008, 883, 129–136 CrossRef PubMed.
- J. J. Shi, Y. H. Liu, R. Guo, X. P. Li, A. Q. He, Y. L. Gao, Y. J. Wei, C. G. Liu, Y. Zhao, Y. Z. Xu, I. Noda and J. G. Wu, Design of a New Concentration Series for the Orthogonal Sample Design Approach and Estimation the Number of Reactions in Chemical Systems, Appl. Spectrosc. DOI:10.1366/14-07759.
- X. P. Li, Q. Bi, S. X. Liu, J. Chen, S. J. Yue, Y. J. Wei, K. Huang, Y. Zhao, H. Z. Liu, Y. J. Zhai, Y. Z. Xu, I. Noda and J. G. Wu, Improvement of the sensitivity of the two-dimensional asynchronous spectroscopy based on the AOSD approach by using a modified reference spectrum, J. Mol. Struct., 2013, 1034, 101–111 CrossRef CAS PubMed.
- X. P. Li, Q. H. Pan, J. Chen, S. X. Liu, A. Q. He, C. G. Liu, Y. J. Wei, K. Huang, L. M. Yang, J. Feng, Y. Zhao, Y. Z. Xu, Y. Ozaki, I. Noda and J. G. Wu, Asynchronous orthogonal sample design scheme for two-dimensional correlation spectroscopy (2D-COS) and its application in probing intermolecular interactions from overlapping infrared (IR) bands, Appl. Spectrosc., 2011, 65, 901–917 CrossRef CAS PubMed.
- X. P. Li, S. X. Liu, J. Chen, S. J. Yue, C. G. Liu, Y. J. Wei, K. Huang, Y. Zhao, Y. Z. Xu, I. Noda and J. G. Wu, The influence of changing the sequence of concentration series on the 2D asynchronous spectroscopy generated by the asynchronous orthogonal sample design (AOSD) approach, Vib. Spectrosc., 2012, 60, 212–216 CrossRef CAS PubMed.
- C. F. Zhang, K. Huang, H. Z. Li, J. Chen, S. X. Liu, Y. Zhao, D. J. Wang, Y. Z. Xu, J. G. Wu, I. Noda and Y. Ozaki, Double orthogonal sample design scheme and corresponding basic patterns in two-dimensional correlation spectra for probing subtle spectral variations caused by intermolecular interactions, J. Phys. Chem. A, 2009, 113, 12142–12156 CrossRef CAS PubMed.
- J. Chen, Q. Bi, S. X. Liu, X. P. Li, Y. H. Liu, Y. J. Zhai, Y. Zhao, L. M. Yang, Y. Z. Xu, I. Noda and J. G. Wu, Double asynchronous orthogonal sample design scheme for probing intermolecular interactions, J. Phys. Chem. A, 2012, 116, 10904–10916 CrossRef CAS PubMed.
- Y. L. Gao, J. Liu, Y. H. Liu, J. J. Shi, S. Weng, L. M. Yang, X. Wen, T. Kang, Y. Z. Xu, I. Noda and J. G. Wu, Characterization of the coordination between Nd3+ and ester groups by using double asynchronous orthogonal sample design approach, J. Mol. Struct., 2014, 1069, 205–210 CrossRef CAS PubMed.
- J. Liu, Y. L. Gao, L. Zheng, D. Gao, A. Q. He, Y. H. Liu, S. Weng, Y. Zhao, Z. Yang, L. M. Yang, X. Wen, Y. Z. Xu, I. Noda and J. G. Wu, Coordination between cobalt(II) ion and carbonyl group in acetone probed by using DAOSD approach, J. Mol. Struct., 2014, 1069, 217–222 CrossRef CAS PubMed.
- Q. Bi, J. Chen, X. P. Li, J. J. Shi, X. Wang, J. Zhang, D. Gao, Y. Zhai, Y. Zhao, S. Weng, Y. Z. Xu, I. Noda and J. G. Wu, Investigation on the dipole-dipole interactions between tetramethylurea and acetonitrile by two-dimensional asynchronous spectroscopy, J. Mol. Struct., 2014, 1069, 264–271 CrossRef CAS PubMed.
- Q. Bi, J. Chen, X. P. Li, J. J. Shi, R. Guo, Y. Zhai, Y. Z. Xu, I. Noda and J. G. Wu, A method based on the DAOSD approach to estimate the variation of the peak position and bandwidth caused by intermolecular interactions, J. Mol. Struct., 2014, 1069, 211–216 CrossRef CAS PubMed.
- Y. L. Gao, D. Li, J. J. Shi, X. M. Wang, T. G. Kang, S. F. Weng, Y. Z. Xu, I. Noda and J. G. Wu, Coordination between lanthanide (III) ions and organic ligands of natural pharmaceutical containing lactone group probed by DAOSD approach, Biomed. Spectrosc. Imaging, 2015, 4, 129–137 CAS.
- Y. H. Liu, J. J. Shi, D. Q. Gao, Y. L. Gao, R. Guo, X. F. Ling, S. F. Weng, Y. Z. Xu, I. Noda and J. G. Wu, Interactions between pyridinium and Nd3+, Chin. Chem. Lett., 2015, 26, 182–186 CrossRef CAS PubMed.
- D. Q. Gao, X. P. Li, J. J. Shi, X. Y. Kang, T. G. Kang, J. M. Xia, X. F. Ling, S. F. Weng, Y. Z. Xu, I. Noda and J. G. Wu, Two-dimensional correlation spectroscopic studies on coordination between carbonyl group of butanone and metal ions, Chin. Chem. Lett., 2015, 26, 177–181 CrossRef CAS PubMed.
- J. Zhang, R. Guo, T. G. Kang, A. Q. He, S. F. Weng, J. M. Xia, X. F. Ling, Y. Z. Xu and J. G. Wu, Spectrosc. Spectral Anal., 2015, 35, 1193–1198 CAS.
- X. P. Li, X. K. Fan, K. Huang, H. Z. Liu, Y. Zhao, Y. J. Wei, C. G. Liu, Y. Z. Xu, I. Noda and J. G. Wu, Characterization of intermolecular interaction between two substances when one substance does not possess any characteristic peak, J. Mol. Struct., 2014, 1069, 127–132 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra16062f |
| This journal is © The Royal Society of Chemistry 2015 |