
β-Cyclodextrin in water: highly facile biomimetic one pot deprotection of phenolic THP/MOM/Ac/Ts ethers and concomitant regioselective cyclization of chalcone epoxides and 2′-aminochalcones†
Sumit Kumar*,
Nishant Verma and
Naseem Ahmed*
Department of Chemistry, Indian Institute of Technology Roorkee, Roorkee 247 667, Uttarakhand, India. E-mail: sumitdcy@iitr.ac.in; nasemfcy@iitr.ac.in; Fax: +91 1332 285745; Tel: +91 1332 285745
First published on 30th September 2015
Abstract
A mild and efficient one-pot deprotection of THP/MOM/Ac/Ts ethers and the concomitant cyclization of chalcone epoxides to 2-hydroxyindanones or 2′-aminochalcones to aza-flavanones using β-cyclodextrin in water has been developed. β-CD was found to be highly effective at carrying out the deprotection and sequential transformations in an eco-friendly environment affording moderate to excellent yields (59–99%) at 60 °C in 8–22 min. Water, an eco-friendly reaction medium, has been utilized for the first time in this reaction. The merits of the presented protocol include the high yields and catalyst reusability and the protocol precludes the use of metals and organic solvents. The present method is much milder but more advanced than those reported earlier.
Introduction
The protection and deprotection of hydroxyl groups have fundamental importance and are frequently used strategies in multi-step organic syntheses. In particular, these two phenomena are extremely important because of the presence of hydroxyl groups in a number of natural products and in biological and synthetic compounds such as carbohydrates, peptides, macrolides, nucleotides, steroids and polyethers.1 Among the various methods for protecting a hydroxy group, the formation of tetrahydropyranyl ethers (THPEs) is the most commonly employed method due to its easy formation and inertness to various reaction conditions, such as catalytic hydrogenation and alkylating or acylating conditions, and to strong bases such as metal hydrides, organolithium compounds and Grignard reagents.2 Likewise, methoxymethyl chloride (MOMCl), acetyl chloride/acetic anhydride (CH3COCl/Ac2O) and tosyl chloride (TsCl) are also important reagents for alcoholic and phenolic group protection. Various methods have been reported for the deprotection of THPEs including the use of protic acids,3a–d BF3-etherate,3e LiBr,3f LiOTf,3g LiBF4,3h LiClO4,3i In(OTf)3,3j Sc(OTf)3,3k I2,3l InCl3,3m ZrCl3,3n CuCl2,3o NH4Cl3p and other catalysts. Similarly, many catalysts have been used to remove the MOM group under acidic conditions such as protic acids,4a Lewis acids,4b Lewis acid–thiol,4c boron halides,4d YbCl3,4e CBr4–PPh3,4f ZnBr2,4g silica-supported NaHSO44h and TMSOTf (TESOTf)-2,2′-bipyridyl.4i Several catalysts have been reported for the deacetylation and detosylation of alcohols and phenols under acidic and basic conditions including NaOMe,5a micelles,5b Zn–MeOH,5c enzymes,5d metallo–enzymes,5e metal complexes,5f antibodies,5g montmorillonite K-10,5h I2,5i NaBO35j and TFA.5k Most of these methods, however, have one or more drawbacks such as low yields, long reaction times, the need to reflux at high temperature, the use of excess amounts of reagents and tedious work-up procedures.6 Hence, there is still scope to develop milder and more efficient methods for the detetrahydropyranylation, demethoxymethylation, deacetylation and detosylation of hydroxyl groups.Similarly, various reagents and reaction conditions have been previously employed for the cyclization of chalcones to aza-flavanones, including acids,7a bases,7b–d silica gel,7e–g light,7h electrolysis,7i nanocrystalline MgO,7j zeolites,7k L-proline,7l Yb(OTf)3,7m,n silica gel supported TaBr5,7o alumina supported-CeCl3·7H2O–NaI,7p and microwave irradiation.7q,r Most of these procedures however have drawbacks, for example low yields, the use of toxic metals and organic solvents and harsh reaction conditions.
In a metal-free green context, cyclodextrins are therefore emerging as alternative catalysts for numerous challenging organic reactions due to the fine tuning of their physico-chemical properties.8–10 Cyclodextrins (CDs), which are cyclic oligosaccharides possessing hydrophobic cavities, exert microenvironmental effects leading to selective reactions. They catalyze reactions by supramolecular catalysis through non-covalent bonding, forming reversible host–guest complexes just like enzymes. We used β-cyclodextrin as the catalyst because it is easily accessible and inexpensive among the CDs. The concept of green chemistry has drawn attention to performing reactions in aqueous media. However, the fundamental problem with performing organic reactions in water is that many organic substrates are hydrophobic and are insoluble in water. However, this can be overcome by the use of cyclodextrins.11
As a continuation of our recent findings concerning the YbCl3-catalyzed ring-opening of epoxides,4e herein, we report a simple and efficient green protocol for the biomimetic one pot deprotection of MOM/THP/Ac/Ts ethers and the concomitant regioselective cyclizations of chalcone epoxides to 2-hydroxyindanones or 2′-aminochalcones to aza-flavanones employing a mild and environmentally friendly β-CD catalyst as a novel reagent in water under microwave conditions. Such 2-hydroxyindanones and aza-flavanones are widely used as synthetic intermediates of high significance in organic and medicinal chemistry.
Results and discussion
In our initial study toward the development of this methodology, a model reaction was conducted by treating a substrate 15 in the presence of various metal salts and then finally β-cyclodextrin. Initially, the catalytic efficiency of different Lewis acids (CdCl2, CoCl2, FeCl3, ZnCl2, SrCl2, CuCl2, NH4Cl, CuI2, InCl3, HgCl2, SnCl2·2H2O and CeCl3) was screened (Table 1).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Mol% | Temp (°C) | Time (min) | Yieldd (%) | ||
| 15a | a | b | |||||
| a Reactions were carried out on a 1 mmol scale.b Anton Paar Monowave-300 reactor®.c H2O was used as the solvent for entries 13–15.d Isolated yields. | |||||||
| 1 | CdCl2 | 10 | 40 | 4 | — | — | 90 |
| 2 | CoCl2 | 10 | 40 | 2 | — | — | 94 |
| 3 | FeCl3 | 10 | 40 | 2 | — | — | 93 |
| 4 | ZnCl2 | 10 | 40 | 2 | — | — | 91 |
| 5 | SrCl2 | 10 | 40 | 10 | — | — | — |
| 6 | CuCl2 | 10 | 40 | 10 | — | 84 | — |
| 7 | NH4Cl | 10 | 40 | 10 | 5 | 55 | — |
| 8 | CuI2 | 10 | 40 | 5 | 20 | 40 | — |
| 9 | InCl3 | 10 | 40 | 5 | 67 | 10 | 20 |
| 10 | HgCl2 | 10 | 40 | 5 | 55 | — | — |
| 11 | SnCl2·2H2O | 10 | 40 | 10 | 58 | — | — |
| 12 | CeCl3 | 5 | 40 | 4 | 75 | — | — |
| 13c | β-CD | 20 | 60 | 10 | 95 | — | — |
| 14c | β-CD | 10 | 60 | 10 | 95 | — | — |
| 15c | β-CD | 5 | 80 | 20 | 65 | — | — |

Metal halides such as CdCl2, CoCl2, FeCl3 and ZnCl2 did not show any catalytic activity in the formation of product 15a, instead they yielded the side product b in excellent yields (Table 1, entries 1–4). SrCl2 was unable to give any of the products (Table 1, entry 5). CuCl2 yielded only the deprotected product a in good yield without cyclization (Table 1, entry 6). Lewis acids like NH4Cl, CuI2 and InCl3 exhibited poor to moderate catalytic activity at 10 mol% catalyst loading furnishing the product 15a in 5–67% yields with the formation of side products a and b also (Table 1, entries 7–9). HgCl2, SnCl2·2H2O and CeCl3 were found to be good catalysts at 10 mol% catalyst loading, which gave 55–75% yields of product 3b without forming any of the side products within 4–10 min (Table 1, entries 10–12). Then, the catalyst optimization was achieved for the β-cyclodextrin catalyst in water.
To optimize the catalyst loading, three consecutive reactions were carried out with 20, 10 and 5 mol% catalyst loadings under microwave irradiation (Table 1, entries 13–15). The reaction proceeded smoothly with β-cyclodextrin in water at 20 and 10 mol% catalyst loadings with the same capacity to provide the product 15a (deprotection with cyclization) in 95% yield (Table 1, entries 13 and 14). Further decreasing the catalyst loading to 5 mol% furnished the product 15a in only 65% yield, even after prolonging the reaction time and temperature (Table 1, entry 15).
We also screened solvents like acetonitrile, CH2Cl2, THF, acetone, DMF, DMSO and neat H2O and solvents like CH2Cl2, THF, acetone, DMF and DMSO gave good results (Table 2, entries 2–6). However, neat H2O was found to give the best results with quantitative conversion (99%) at 60 °C under microwave irradiation. Despite the excellent results, we further explored other reaction conditions for the above reaction in order to target the optimum protocol for the transformation. For example, the reaction was also subjected to conventional heating, however this resulted in a 81% yield of the product (Table 2, entry 9) in 5 h. The advantage of the use of microwave irradiation over conventional heating was that it helped reduce the reaction time from hours to minutes. Hence, the optimum conversion of the substrate 15 to the product 15a was attained when the reaction was carried out utilising 10 mol% β-cyclodextrin in water at 60 °C for 10 min under microwave condition.

Then, under the optimal reaction conditions, we carried out the deprotections of various phenolic and alcoholic THP/MOM/Ac/Ts ethers and the concomitant ring openings of the chalcone epoxides using β-cyclodextrin in water (Table 3). The reactions underwent deprotection and sequential cyclization in excellent yields for detetrahydropyranylation, demethoxymethylation and deacetylation (92–99%) but in moderate yields for detosylation (61–74%) within 8–9 min at 60 °C (Table 3, entries 1–9). In the case of the chalcones (5–14), only deprotection was observed without cyclization. Interestingly, deprotection of the THP/MOM ethers followed by Friedel–Crafts alkylation was observed for the chalcone epoxides (15–21) giving the corresponding 2-hydroxyindanones (15a–21a) in excellent yields (95–99%) within 10 min. We also subjected alicyclic tetrahydropyranyl tosylates (22–26) to the same reaction protocol, which resulted in moderate yields (59–63%) of their respective products (22a–26a) in 14–15 min. All the synthesized products were characterized by mass spectrometry, FTIR, NMR and also by comparison with the available literature.4e,5k
| Entry | Substrate | Product | Time (min) | Yields (%) | |||
|---|---|---|---|---|---|---|---|
| ab | bc | cd | de | ||||
| a Reactions were carried out on a 1 mmol scale at 60 °C under MWI with β-CD (10 mol%).b Isolated yields from OTHP.c Isolated yields from OMOM.d Isolated yields from OAc.e Isolated yields from OTs. | |||||||
| 1 |  |
 |
8 | 99 | 99 | 99 | 73 |
| 2 |  |
 |
9 | 99 | 99 | 99 | 70 |
| 3 |  |
 |
8 | 99 | 98 | — | — |
| 4 |  |
 |
8 | 98 | 97 | 99 | 61 |
| 5 |  |
 |
8 | 95 | 94 | 97 | 68 |
| 6 |  |
 |
8 | 95 | 94 | 98 | 72 |
| 7 |  |
 |
8 | 96 | 95 | 94 | 70 |
| 8 |  |
 |
8 | 93 | 95 | 94 | 67 |
| 9 |  |
 |
8 | 95 | 94 | 97 | 69 |
| 10 |  |
 |
8 | 93 | 92 | 97 | 70 |
| 11 |  |
 |
8 | 95 | 93 | 95 | 72 |
| 12 |  |
 |
8 | 96 | 92 | 93 | 72 |
| 13 |  |
 |
8 | 93 | 97 | 94 | 74 |
| 14 |  |
 |
8 | 95 | 98 | 97 | 71 |
| 15 |  |
 |
10 | 99 | 99 | — | — |
| 16 |  |
 |
10 | 99 | 98 | — | — |
| 17 |  |
 |
10 | 98 | 99 | — | — |
| 18 |  |
 |
10 | 96 | 97 | — | — |
| 19 |  |
 |
10 | 98 | 95 | — | — |
| 20 |  |
 |
10 | 96 | 95 | — | — |
| 21 |  |
 |
10 | 99 | 97 | — | — |
| 22 |  |
 |
14 | — | — | — | 62 |
| 23 |  |
 |
14 | — | — | — | 60 |
| 24 |  |
 |
14 | — | — | — | 61 |
| 25 | 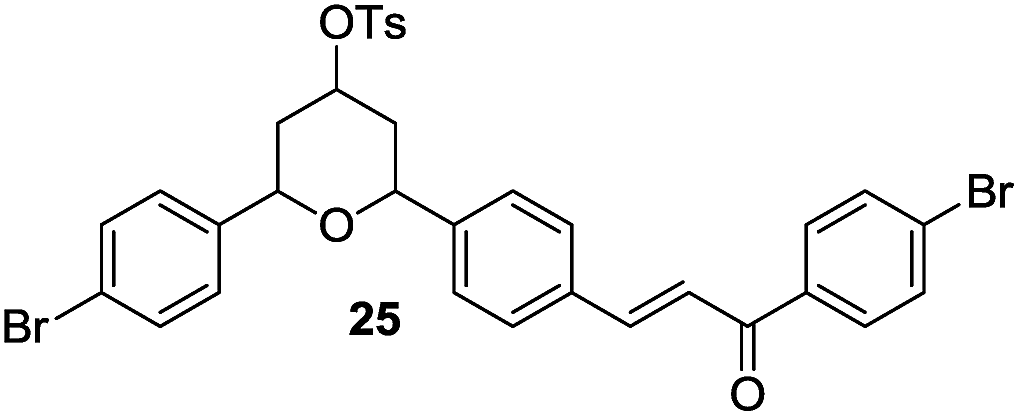 |
 |
15 | — | — | — | 59 |
| 26 |  |
 |
15 | — | — | — | 63 |
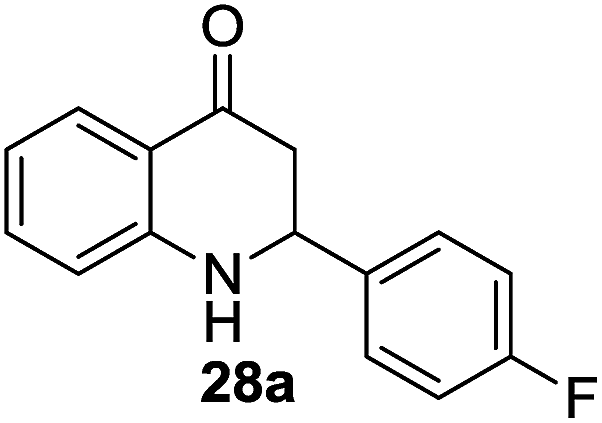
The scope of the β-cyclodextrin catalyst was also examined for the cyclization of 2′-aminochalcones (27–36) to their corresponding aza-flavanones (27a–36a). Under the optimal reaction conditions, β-cyclodextrin catalyzed the isomerization of 2′-aminochalcones to their corresponding aza-flavanones in excellent yields (70–96%) within 9–22 min (Table 4).
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time (min) | Yield (%) |
| a Reactions were carried out on 1 mmol scale at 60 °C under MWI, β-CD (10 mol%).b Isolated yields from OTHP.c Isolated yields from OMOM.d Isolated yields from OAc.e Isolated yields from OTs. | ||||
| 1 |  |
 |
15 | 96 |
| 2 |  |
 |
20 | 90 |
| 3 |  |
 |
18 | 92 |
| 4 |  |
 |
20 | 93b, 92c, 95d, 70e |
| 5 |  |
 |
20 | 88b, 90c, 94d,73e |
| 6 |  |
 |
10 | 96 |
| 7 |  |
 |
9 | 95 |
| 8 |  |
 |
9 | 93 |
| 9 |  |
 |
22 | 81 |
| 10 |  |
 |
12 | 89 |

In addition, we also studied the substituent effects of electron-donating to electron-withdrawing groups on the Ar2 ring of the 2′-aminochalcones on the reaction rate and product yields. As anticipated, electron donating groups enhanced the cyclization to give the corresponding aza-flavanones in good yields, while electron withdrawing groups slow down the cyclization leading to moderate yields of aza-flavanones. For example, electron-donating groups such as –CH3, –OCH3 and –SCH3 groups on Ar2 afforded the products in excellent yields at a faster rate (Table 4; 32a–34a, 36a). Substituents such as –F and –Cl on Ar2 furnished the corresponding products in excellent yields (90–92%) but after a longer reaction time (Table 4; 28a, 29a). Similarly, electron-withdrawing groups, such as –NO2, on Ar2 gave the product in 81% yield also with a longer reaction time (Table 4; 35a). In the case of the cyclization of 2′-hydroxychalcones to their corresponding oxa-flavanones, the reactions didn’t proceed and this might be due to the poor nucleophilicity of the hydroxyl groups under similar reaction conditions.
All of the structures of the isolated products were confirmed by IR and 1H NMR spectral analysis and from available literature data.7o,p For example, the 1H-NMR spectrum of 2-(3,4-dimethoxyphenyl)-2,3-dihydroquinolin-4(1H)-one 33a shows a doublet of doublets at 5.43 ppm due to –CH proton at 2-position and two doublets of doublets (dd) at 2.88–3.13 ppm due to the –CH2 protons at the 3-position, which are characteristic of flavanones, and a peak for one proton at 12.17 ppm, due to the –NH is in agreement with the cyclization of the 2′-aminochalcone to its aza-flavanone.
A plausible reaction mechanism for the THP ether deprotection and the simultaneous intramolecular Friedel–Crafts alkylation is depicted in Scheme 1. The role of β-CD appears to be not only to activate the THP ether and epoxide by hydrogen bonding, but also to promote the highly regioselective ring opening from the β-carbon due to the considerable electron deficient character at the benzylic position rather than from the α-carbon due, which would result in a 4-membered cyclobutanone (unstable intermediate, route-2) (Fig. 1). Therefore, route 1 was preferred to route 2 because of the stabilised carbocation formation and nucleophilic attack took place at the β-carbon because of the better resonance stabilized benzyl intermediate, which resulted in faster epoxide ring opening from the β-carbon. This lead to a five membered ring, which on subsequent hydrolysis gave the desired product with concurrent liberation of β-CD for next catalytic cycle. In these reactions, β-CD was recycled and reused.
 | ||
| Scheme 1 Plausible reaction mechanism for the β-cyclodextrin induced deprotection of a phenolic THP ether and concomitant intramolecular Friedel–Crafts alkylation. | ||
 | ||
| Fig. 1 Mechanistic rationale for the β-cyclodextrin induced deprotection of a phenolic ether and concomitant cyclization. | ||
Conclusions
Thus, we have reported for the first time the successful use of β-cyclodextrin in water for the deprotection of THP/MOM/Ac/Ts ethers and the concomitant regioselective ring opening of chalcone epoxides via intramolecular Friedel–Crafts alkylation to produce 2-hydroxyindanones or the cyclization of 2′-aminochalcones to aza-flavanones. Such supramolecular catalysis afforded the products in excellent yields (59–99%) within 8–22 min and therefore represents a novel methodology. The high regioselectivity was achieved due to the formation of inclusion complexes between the epoxides and β-CD. This method precludes the use of organic solvents, toxic metals and acid or base catalysts. Some important features of the method include the high yields, shorter reaction times, green conditions, reusability of the catalyst and easy workup.Experimental
All the required chemicals were purchased from the Merck and Aldrich Chemical Companies. Pre-coated aluminium sheets (silica gel 60 F254, Merck) were used for thin-layer chromatography (TLC) and spots were visualized under UV light. Silica gel column chromatography was performed using silica gel 60–120 mesh size (RANKEM Limited). IR spectra were recorded with KBr on a Thermo Nicolet FT-IR spectrophotometer. 1H NMR and 13C NMR spectra were recorded on Jeol ECX 400 MHz and Bruker Spectrospin DPX 500 MHz spectrometers using CDCl3 as the solvent and trimethylsilane (TMS) as the internal standard. Spectra were processed using Bruker Topspin® 3.0.b.8 software. Splitting patterns were designated as follows; s = singlet, d = doublet, dd = doublet of doublets, m = multiplet, br = broad. Chemical shift (δ) values are given in ppm. Mass spectra were collected using a direct inlet system (70 eV) with a VL detector (ES, 4000 V) on a Perkin Elmer GC-MS. High-resolution mass spectra (HRMS) were obtained on a Brüker microTOF™-Q II mass spectrometer (ESIMS).Microwave irradiation experiments
All microwave experiments were carried out in a dedicated Anton Paar Monowave-300 reactor®, operating at a frequency of 2.455 GHz with continuous irradiation power of 0 to 850 W. The reactions were performed in G-30 borosilicate glass vials sealed with Teflon septa and placed in the microwave cavity. Initially, microwave irradiation of a required power was used and the temperature was ramped from room temperature to a desired temperature. Once this temperature was attained, the process vial was held at this temperature for the required time. The reactions were continuously stirred. The temperature was measured by an IR sensor. After the experiments a cooling jet cooled the reaction vessel to ambient temperature.General procedure for the microwave-assisted deprotection of THP, MOM, acetyl and tosyl ethers and concomitant cyclization of the chalcone epoxides and 2′-aminochalcones
The substrate (1 mmol) dissolved in water (2 mL) was added to an aqueous solution of β-cyclodextrin (10 mol% in 10 mL of water) kept in a G-30 process vial and capped with a Teflon septum. After pre-stirring for one minute, the vial was subjected to microwave irradiation with the holding temperature of 60 °C for the prescribed time (Tables 3 and 4). After completion of the reaction, the mixture was cooled to room temperature and extracted with EtOAc (3 × 15 mL) and the catalyst was filtered off and washed with EtOAc (2 × 10 mL), the filtrate was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as an eluent if required, otherwise compounds were pure enough for spectral elucidation.
:2) as an eluent if required, otherwise compounds were pure enough for spectral elucidation.
Characterization data for representative compounds
Acknowledgements
The authors wish to express their gratitude to the BRNS, India for financial support, DST, New Delhi for providing HRMS facility and CSIR, New Delhi for awarding SRF to S. K.Notes and references
- (a) V. Amaranth and A. D. Broom, Chem. Rev., 1977, 77, 183 CrossRef; (b) D. N. Robertson, J. Org. Chem., 1960, 25, 931 CrossRef CAS.
- A. R. Hajipour, M. Kargosha and A. E. Ruoho, Synth. Commun., 2009, 39, 1084 CrossRef CAS PubMed.
- (a) B. Tamami and K. Parvanak, Tetrahedron Lett., 2004, 45, 715 CrossRef CAS PubMed; (b) V. V. Namboodiri and R. S. Varma, Tetrahedron Lett., 2002, 43, 1143 CrossRef CAS; (c) B. S. Babu and K. Balasubramanian, Tetrahedron Lett., 1998, 39, 9287 CrossRef CAS; (d) P. N. Reddy, B. K. Sunil, P. S. Kumar, N. Y. Srinivasulu, T. Reddy and B. Rajitha, Chem. Heterocycl. Compd., 2005, 41, 11 CrossRef; (e) R. R. Diaz, C. R. Melgarejo, M. T. Plaza and I. I. Cubero, J. Org. Chem., 1994, 59, 7928 CrossRef; (f) M. Narender, M. S. Reddy and K. R. Rao, Synthesis, 2004, 30, 1741 Search PubMed; (g) C. Wiles, P. Watts and S. Haswell, Tetrahedron Lett., 2005, 61, 5209 CrossRef CAS PubMed; (h) B. S. Babu and K. K. Balasubramanian, Tetrahedron Lett., 1998, 39, 9287 CrossRef CAS; (i) B. Karimi and J. Maleski, Tetrahedron Lett., 2002, 43, 5353 CrossRef CAS; (j) B. C. Ranu and M. Saha, J. Org. Chem., 1994, 59, 8269 CrossRef CAS; (k) T. Mineno, Tetrahedron Lett., 2002, 43, 7975 CrossRef CAS; (l) G. G. Haraldsson and J. E. Baldwin, Tetrahedron, 1997, 53, 215 CrossRef CAS; (m) K. Tanemura, T. Haraguchi and T. Suzuki, Bull. Chem. Soc. Jpn., 1992, 65, 304 CrossRef CAS; (n) H. M. S. Kumar, B. V. S. Reddy, E. J. Reddy and J. S. Yadav, Chem. Lett., 1999, 28, 857 CrossRef; (o) J. S. Yadav, D. Srinivas and G. S. Reddy, Synth. Commun., 1998, 28, 1399 CrossRef CAS PubMed; (p) A. T. B. Molnarand, Tetrahedron Lett., 1996, 37, 8597 CrossRef.
- (a) S. Amano, N. Takemura, M. Ohtsuka, S. Ogawa and N. Chida, Tetrahedron, 1999, 55, 3855 CrossRef CAS; (b) G. V. M. Sharma, K. L. Reddy, P. S. Lakshmi and P. R. Krishna, Tetrahedron Lett., 2004, 45, 9229 CrossRef CAS PubMed; (c) G. R. Kieczykowski and R. H. Schlessinger, J. Am. Chem. Soc., 1978, 100, 1938 CrossRef CAS; (d) L. A. Paquette, Z. Gao, Z. Ni and G. F. Smith, Tetrahedron Lett., 1997, 38, 1271 CrossRef CAS; (e) S. Kumar, N. Verma, I. Parveen and N. Ahmed, J. Heterocycl. Chem., 2015 DOI:10.1002/jhet.2517; (f) Y. Peng, C. Ji, Y. Chen, C. Huang and Y. Jiang, Synth. Commun., 2004, 34, 4325 CrossRef CAS PubMed; (g) J. H. Hana, Y. E. Kwona, J.-H. Sohnb and D. H. Ryua, Tetrahedron, 2010, 66, 1673 CrossRef PubMed; (h) C. Ramesh, N. Ravindranath and B. Das, J. Org. Chem., 2003, 68, 7101 CrossRef CAS PubMed; (i) H. Fujioka, O. Kubo, K. Senami, Y. Minamitsuji and T. Maegawa, Chem. Commun., 2009, 4429 RSC.
- (a) J. Otera, Chem. Rev., 1993, 93, 1449 CrossRef CAS; (b) T. Kunitake, Y. Okahata and T. Sakamoto, J. Am. Chem. Soc., 1976, 98, 7799 CrossRef CAS; (c) A. G. Gonzalez, Z. D. Jorge, H. L. Dorta and F. L. Rodriguez, Tetrahedron Lett., 1981, 22, 335 CrossRef CAS; (d) V. S. Parmer, A. K. Prasad, N. K. Sharma, K. S. Bisht, H. N. Pati and P. Taneja, Bioorg. Med. Chem. Lett., 1993, 3, 585 CrossRef; (e) M. R. Crampton, K. E. Holt and J. M. Perey, J. Chem. Soc., 1990, 2, 1701 Search PubMed; (f) V. L. Boisselier, M. Postel and E. Dunach, Tetrahedron Lett., 1997, 38, 2981 CrossRef; (g) J. Guo, W. Huang and T. S. Scanlan, J. Am. Chem. Soc., 1994, 116, 8062 Search PubMed; (h) B. P. Bandgar, L. S. Uppalla, A. D. Sagar and V. S. Sadavarte, Tetrahedron Lett., 2001, 42, 1163 CrossRef CAS; (i) B. Das, J. Banerjee, R. Ramu, R. Pal, N. Ravindranath and C. Ramesh, Tetrahedron Lett., 2003, 44, 5465 CrossRef CAS; (j) B. P. Bandgar, L. S. Uppalla and V. S. S. Patil, New J. Chem., 2003, 5, 68 Search PubMed; (k) N. Ahmed, N. K. Konduru, S. Ahmad and M. Owais, Eur. J. Med. Chem., 2014, 75, 233 CrossRef CAS PubMed.
- L. Liu, M. Chen and C. Cai, Chin. Chem. Lett., 1992, 8, 585 Search PubMed.
- (a) P. Kulkarni, P. Wagh and P. Zubaidha, J. Chem., 2012, 2, 106 CAS; (b) A. Aitmambetov and A. Kubzheterova, Russ. J. Bioorg. Chem., 2002, 28, 165 CrossRef CAS; (c) K. Tanka and T. Sugino, Green Chem., 2001, 3, 133 RSC; (d) R. Mondal, A. D. Gupta and A. K. Mallik, Tetrahedron Lett., 2011, 52, 5020 CrossRef CAS PubMed; (e) N. Ahmed, H. Ali and J. E. van Lier, Tetrahedron Lett., 2005, 46, 253 CrossRef CAS PubMed; (f) G. J. Sagrerai and G. A. Seoane, J. Braz. Chem. Soc., 2005, 16, 851 CrossRef; (g) J. J. Li, L. Y. Jin, C. M. Yu and W. K. Su, J. Chem. Res., 2009, 3, 170 CrossRef; (h) Y. Maki, K. Shimada, M. Sakoand and K. Hirota, Tetrahedron, 1988, 44, 3187 CrossRef CAS; (i) Z. Sanicanin and I. Tabakovic, Tetrahedron Lett., 1986, 27, 407 CrossRef CAS; (j) B. Choudary, K. V. S. Ranganath, J. Yadav and M. L. Kantam, Tetrahedron Lett., 2005, 46, 1369 CrossRef CAS PubMed; (k) M. J. Climent, A. Corma, S. Iborra and J. J. Primo, J. Catal., 1995, 151, 60 CrossRef CAS; (l) S. Chandrasekhar, K. Vijeender and K. V. Reddy, Tetrahedron Lett., 2005, 46, 6991 CrossRef CAS PubMed; (m) V. K. Rao, M. S. Rao and A. Kumar, J. Heterocycl. Chem., 2011, 48, 1356 CrossRef CAS PubMed; (n) S. Genovese, S. Fiorito, M. C. Specchiulli, V. A. Taddeo and F. Epifano, Tetrahedron Lett., 2015, 56, 847 CrossRef CAS PubMed; (o) N. Ahmed and J. E. van Lier, Tetrahedron Lett., 2006, 47, 2725 CrossRef CAS PubMed; (p) N. Ahmed and J. E. van Lier, Tetrahedron Lett., 2007, 48, 13 CrossRef CAS PubMed; (q) T. Patonay, R. S. Varma, A. Vass, A. Levai and J. Dudas, Tetrahedron Lett., 2001, 42, 1403 CrossRef CAS; (r) G. W. Kabalka and A. R. Mereddy, Tetrahedron Lett., 2005, 46, 6315 CrossRef CAS PubMed.
- (a) M. A. Reddy, K. Surendra, N. Bhanumathi and K. R. Rao, Tetrahedron, 2002, 58, 6003 CrossRef CAS; (b) N. S. Krishnaveni, K. Surendra, Y. V. D. Nageswar and K. R. Rao, Synthesis, 2003, 13, 1968 Search PubMed; (c) M. Narender, M. S. Reddy and K. R. Rao, Synthesis, 2004, 11, 1741 Search PubMed.
- (a) K. Surendra, N. S. Krishnaveni, M. A. Reddy, Y. V. D. Nageswar and K. R. Rao, J. Org. Chem., 2003, 68, 2058 CrossRef CAS PubMed; (b) K. Surendra, N. S. Krishnaveni, M. A. Reddy, Y. V. D. Nageswar and K. R. Rao, J. Org. Chem., 2003, 68, 9119 CrossRef CAS PubMed; (c) M. A. Reddy, N. Bhanumathi and K. R. Rao, Chem. Commun., 2001, 1974 RSC.
- (a) L. R. Reddy, N. Bhanumathi and K. R. Rao, Chem. Commun., 2000, 2321 RSC; (b) M. A. Reddy, L. R. Reddy, N. Bhanumathi and K. R. Rao, New J. Chem., 2001, 25, 359 RSC; (c) S. N. Murthy and Y. V. D. Nageswar, Tetrahedron Lett., 2011, 52, 4481 CrossRef PubMed; (d) K. Surendra, N. S. Krishnaveni, R. Sridhar and K. R. Rao, Tetrahedron Lett., 2006, 47, 2125 CrossRef CAS PubMed.
- (a) S. N. Murthy and Y. V. D. Nageswar, Tetrahedron Lett., 2011, 52, 4481 CrossRef PubMed; (b) O. S. Tee, C. Mazza, R. L. Hemmer and J. B. Giorgi, J. Org. Chem., 1994, 59, 7602 CrossRef CAS; (c) S. Kumar and N. Ahmed, Green Chem., 2015 10.1039/c5gc01785h; (d) S. Kumar and N. Ahmed, RSC Adv., 2015, 5, 77075 RSC; (e) N. Verma, V. Kundi and N. Ahmed, Tetrahedron Lett., 2015, 28, 4175 CrossRef PubMed; (f) S. Kumar, N. Konduru, N. Verma and N. Ahmed, Synth. Commun., 2015 DOI:10.1080/00397911.2015.1093142.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra15996b |
| This journal is © The Royal Society of Chemistry 2015 |