
Enhancement of icephobic properties based on UV-curable fluorosilicone copolymer films†
Abstract
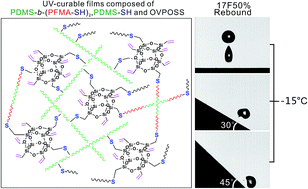
UV-curable films composed of thiol-terminated fluorosilicone methacrylate triblock copolymers (PDMS-b-(PFMA-SH)2), thiol-functionalized PDMS (PDMS-SH) and octavinyl POSS (OVPOSS) were developed for the purpose of icephobic application. The PDMS-b-(PFMA-SH)2 copolymers were firstly synthesized via reversible addition–fragmentation chain transfer polymerization of dodecafluoroheptyl methacrylate (12FMA) or 2-perfluorooctylethyl methacrylate (17FMA), and followed by thiol-modification of the end groups. The results of differential scanning calorimetry and X-ray diffraction revealed the presence of perfluoroalkyl side groups in P17FMA as presented in the crystalline structure. These endowed the P17FMA-containing films with higher receding contact angles (100.2–113.5°) as compared with the P12FMA-containing films. In icephobicity investigations of the prepared films, it was found that the rebound of the impacting droplet was influenced by the receding contact angle, surface roughness, and temperature of the film surface. Water droplets could rebound from the horizontal and tilted (30° and 45°) P17FMA-containing film surfaces down to −15 °C, allowing surface dewetting before the water droplets froze, while the droplets adhered to the P12FMA-containing film surfaces. Moreover, the ice shear strengths of all the prepared film surfaces were lower than 210 kPa, only about 15% of the value on the bare aluminum surface. Therefore, P17FMA-containing UV-curable films could be a potential candidate for icephobic applications.
 Please wait while we load your content...
Please wait while we load your content...

