
Pd/C-catalyzed facile synthesis of primary aromatic amides by aminocarbonylation of aryl iodides using ammonia surrogates†
Rajendra S. Mane and
Bhalchandra M. Bhanage*
Department of Chemistry, Institute of Chemical Technology, N. Parekh Marg, Matunga, Mumbai- 400 019, India. E-mail: bm.bhanage@gmail.com; bm.bhanage@ictmumbai.edu.in; Fax: +91 22 33611020; Tel: +91 22 33612601
First published on 3rd September 2015
Abstract
Use of Pd/C as an efficient, phosphine free, heterogeneous and recyclable catalyst for one pot carbonylative synthesis of primary aromatic amides has been demonstrated. The developed protocol is simple to perform and employs ammonium carbamate as an in situ solid ammonia source which is feasible and more convenient than direct use of ammonia gas. The applicability of the developed protocol was studied for various aryl iodide substrates and it was found that it provides good to excellent yields of the desired amides. The catalyst is easily separable and shows significant recyclability for up to four consecutive cycles without loss of catalytic activity.
Introduction
Amides form the elemental backbone of biological molecules and amide chemistry is important for the synthesis of various pharmaceutical active intermediates.1 They play a vital role in the synthesis of peptides, proteins, natural products, agrochemicals, materials, heterocycles, intensifiers of perfumes, anti-block reagents, colour pigments for inks, detergents and lubricants.2 Primary amides can be transformed into a variety of functional groups like amines,3 nitriles,4 secondary and tertiary amides.5 Generally, the primary amides are synthesized by oxidation of benzyl amines,6 benzyl alcohol and benzaldehydes7 or by the reaction of acids, esters, anhydrides and acyl chlorides with ammonia.8 Several research groups have reported partial hydrolysis of nitriles for synthesis of primary amides.9 However, these protocols suffer from several drawbacks such as use of stoichiometric reagents, expensive and hazardous starting materials, poor atom efficiency, harsh reaction conditions and tedious work up.The aminocarbonylation of aryl iodides by using transition metal catalysts has been reported for the synthesis of aromatic amides.10 Palladium-catalyzed carbonylation of aryl iodides as an efficient synthetic route for synthesis of aromatic carbonyl compounds11 which are the essential building blocks in organic synthesis. Literature survey reveals that very few reports are known for synthesis of primary amides as compared to secondary and tertiary amides. In this context, Beller and co-workers have reported palladium catalyzed aminocarbonylation of aryl iodides using ammonia gas and moisture sensitive phosphine ligands for synthesis of primary amides is an undesirable.12 Besides that, the lower nucleophilicity, corrosiveness, hazardous nature and difficulty in handling of NH3 gas during the course of reaction makes the process inconvenient. Moreover, variety of surrogates have been employed including hexamethyldisilazane (HMDS),13 formamide,14 N-tert-butylamides,15 hydroxylamine,16 and even a titanium–nitrogen complex17 to get rid of the inconvenience caused by direct use of ammonia gas. On the similar lines it is worth mentioning that Skrydstrup and Skoda-Foldes have reported the synthesis of primary aromatic amides using homogenous palladium precursors with phosphine ligands.18 In 2013, Alper et al. have reported Pd(OAc)2/CYTOP®292 catalyzed aminocarbonylation of aryl iodides using aqueous ammonia.19 Recently, we also have demonstrated PdCl2 catalysed carbonylation by utilizing methoxylamine hydrochloride as an ammonia source.20 However, despite of their potential utility, the above methodologies suffer from various drawbacks such as two step reactions, use of hazardous, highly flammable HMDS as a ammonia source, use of expensive and moisture sensitive phosphine ligands with harsh reaction conditions, homogenous and non-recyclable precious metal precursors etc.
Homogeneous protocols using phosphine ligands are highly expensive and moisture sensitive, difficult to separate after reaction and throwing it without recycled is not an economical process. To steer clear the deficiencies associated with non-recyclable homogeneous catalysts, use of heterogeneous catalysts is being viewed as an environmentally benign and economically viable alternative. The major advantage of heterogeneous catalysts is, it can be easily separable and possess significant recyclability.
Recently, several groups including ours have reported phosphine-free carbonylation reactions using Pd/C as an efficient heterogeneous, recyclable and environmentally benign catalyst.21 Based on this point of view, herein, we report the Pd/C as an efficient heterogeneous, phosphine free and recyclable catalyst for aminocarbonylation of aryl iodides (Scheme 1). The present protocol simple to operate and makes use of ammonium carbamate as an solid ammonia surrogate it produces two moles of ammonia after mild heating, which is greener and efficient source of ammonia than traditionally used.22
 | ||
| Scheme 1 Pd/C catalysed aminocarbonylation of aryl iodides. | ||
Results and discussion
Our study begun with the reaction of 4-iodotoluene 1a (1 mmol) with ammonium carbonate (1.5 mmol) as an ammonia surrogate, DABCO (2 mmol) using 10% Pd/C (5 mol%), KI (0.5 mmol) under CO atmosphere in 10 mL MeCN at 90 °C for 8 h. The 70% yield of desired product 3a was obtained (Table 1, entry 1). To our delight, various ammonia sources were screened as shown in Table 1 for the model reaction at parameters optimized in the preceding discussion including NH4HCO3, aq. NH3, NH4Cl, CH3CO2NH4, HCO2NH4 and NH2CO2NH4. Among these, ammonium carbamate was found to be the best ammonia source and gave 92% yield of the 3a (Table 1, entry 8). It was observed that use of ammonium carbonate or ammonium bicarbonate results in lowering the yield of desired primary amides (Table 1, entries 1 & 2). Next, we studied the effect of loading of ammonia source and 1.5 mmol of ammonium carbamate gives the highest yield of 3a.| Entry | Ammonia sources | Yieldb (%) |
|---|---|---|
| a Reaction conditions: 1a (1 mmol), ammonia equivalent (1.5 mmol), 2.5 mol% Pd/C (10%), KI (0.5 mmol), DABCO (2 mmol), MeCN (10 mL) under CO (10 atm) at 90 °C, for 8 h.b Yield determined by GC.c Reaction were carried out at 15 atm CO.d Ammonium carbamate (1 mmol). | ||
| 1 | (NH4)2CO3 | 70 |
| 2 | NH4HCO3 | 65 |
| 3 | aq. NH3 | 55 |
| 4 | NH4Cl | 63 |
| 5c | NH4Cl | 70 |
| 6 | CH3CO2NH4 | 20 |
| 7 | HCO2NH4 | 10 |
| 8 | NH2CO2NH4 | 92 |
| 9d | NH2CO2NH4 | 78 |
Increase in the amount of ammonium carbamate had no significant effect on the yield of 3a. While decrease in the amount of ammonium carbamate results into lowering the yield (Table 1, entry 9).
In the next set of experiments, we investigated effect of different reaction parameters such as catalyst and loading of catalyst, solvents, bases, iodide promoters, Pd to iodide ratio, temperature and CO pressure on the model reaction. The results are summarized in Table 2. At first, we screened 5% Pd/C and 10% Pd/C catalyst and their loading for this transformation. Compared with 5% Pd/C, it was found that 10% Pd/C (2.5 mol%) gives highest yield of the desired product 3a (Fig. 1). Decreasing the catalyst loading resulted into decrease in the yield of 3a. However, in the absence of catalyst there was no product formation, indicating that catalyst is solely responsible for the reaction. Also, the effect of different solvents on yield of reaction were investigated such as 1,4-dioxane, toluene, THF, DMF, DMSO and MeCN (Table 2, entries 1–6). Acetonitrile was found to be the best solvent providing an excellent yield of 3a. Further, effect of additives such as KI, NaI, TBAI and TBAB on catalytic activity of the catalyst was studied. The observation concludes that yield of desired product with KI and NaI are the highest and comparable to each other (Table 2, entry 2 and 9). When the reaction was performed in the absence of KI low yield of 3a was obtained (Table 2, entry 12). Furthermore, the influence of Pd to additives ratio was tested. It was noted that yield of desired product increases with decrease in Pd/KI ratio. Thus the optimized catalyst ratio was found to be 2.5 mol% Pd/C with 0.5 mmol KI which offers maximum yield of 3a (Table 2, entry 2). This suggests that addition of KI significantly enhances the catalytic activity as well as yield of the reaction. Various organic and inorganic bases were studied for aminocarbonylation of aryl iodides and the results showed that organic bases such as DABCO and Et3N provide an excellent yield of the product 3a (Table 2, entry 2 and 13) as compared to the inorganic bases. Use of 2.0 mmol of base proved to be essential affording 92% yield of the desired product 3a, and decreasing the amount of base below to 2.0 mmol resulted into lowering the yield.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Additives | Base | T (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (1 mmol), ammonium carbamate (1.5 mmol), Pd/C (2.5 mol%), KI (0.5 mmol), base (2 mmol), solvent (10 mL) under CO (10 atm) for 8 h.b Yield determined by GC.c KI (0.3 mmol).d KI (1 mmol).e Base (1.5 mmol).f Reactions were carried out at 5 and 15 atm of CO.g Reactions were carried out at 5 and 15 atm of CO.h 1,4-diazabicyclo[2.2.2]octane. | ||||||
| Effect of solvent | ||||||
| 1 | 1,4 dioxane | KI | DABCOh | 90 | 8 | 30 |
| 2 | MeCN | KI | DABCO | 90 | 8 | 92 |
| 3 | THF | KI | DABCO | 90 | 8 | 55 |
| 4 | Toluene | KI | DABCO | 90 | 8 | 76 |
| 5 | DMF | KI | DABCO | 90 | 8 | 47 |
| 6 | DMSO | KI | DABCO | 90 | 8 | Trace |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Effect of additive | ||||||
| 7c | MeCN | KI | DABCO | 90 | 8 | 81 |
| 8d | MeCN | KI | DABCO | 90 | 8 | 92 |
| 9 | MeCN | NaI | DABCO | 90 | 8 | 88 |
| 10 | MeCN | TBAI | DABCO | 90 | 8 | 53 |
| 11 | MeCN | TBAB | DABCO | 90 | 8 | 57 |
| 12 | MeCN | — | DABCO | 90 | 8 | 70 |
|
||||||
| Effect of base | ||||||
| 13 | MeCN | KI | Et3N | 90 | 8 | 85 |
| 14 | MeCN | KI | NaHCO3 | 90 | 8 | 56 |
| 15 | MeCN | KI | Na2CO3 | 90 | 8 | 46 |
| 16 | MeCN | KI | K2CO3 | 90 | 8 | 64 |
| 17 | MeCN | KI | DBU | 90 | 8 | Trace |
| 18 | MeCN | KI | NaOAc | 90 | 8 | 49 |
| 19e | MeCN | KI | DABCO | 90 | 8 | 78 |
|
||||||
| Effect of temperature, time and CO pressure | ||||||
| 20 | MeCN | KI | DABCO | 70 | 8 | 68 |
| 21 | MeCN | KI | DABCO | 80 | 8 | 82 |
| 22 | MeCN | KI | DABCO | 90 | 6 | 73 |
| 23 | MeCN | KI | DABCO | 90 | 12 | 94 |
| 24f | MeCN | KI | DABCO | 90 | 8 | 73 |
| 25g | MeCN | KI | DABCO | 90 | 8 | 93 |

 | ||
| Fig. 1 Effect of the Pd/C loading for the aminocarbonylation reaction. | ||
Finally, the effect of temperature, effect of CO pressure and time for the effective progress of the reaction was studied. The temperature study revealed that the yield of 3a increases with increase in the reaction temperature from 70 to 90 °C (Table 2, entries 2, 20 and 21). Further increase in reaction temperature to 100 °C, we does not observed any significant effect on the yield of product 3a. Thus, the temperature 90 °C was selected for further optimization study. The yield of 3a increases when time span of reaction increases from 6 h to 8 h. While, reaction time increases beyond 8 h had no significant effect on yield of product (Table 2, entry 23). Thus, the time study indicates that 8 h is an optimal time for the reaction (Table 2, entry 2). It can also be noted that decrease in CO pressure from 10 to 5 atm decreases the yield of 3a, whereas further increase in CO pressure above 10 atm had no significant effect on the yield of desired product (Table 2, entries 2, 24, 25). Thus, the optimized reaction conditions are as follows: aryl iodide (1 mmol), ammonium carbamate (1.5 mmol), Pd/C (2.5 mol%), KI (0.5 mmol), DABCO (2 mmol) in 10 mL MeCN under 10 atm CO pressure at 90 °C for 8 h.
With these optimized reaction conditions in hand, we checked the effect of various substituents on aryl iodide for broaden the general applicability of developed protocol. Aryl iodides bearing electron-donating substituents such as 2-Me, 4-Me, 2-OMe and 4-OMe result into an excellent yield of corresponding products (Table 3, entries 1–4). The carbonylation of iodobenzene and 1-iodonapthalene was tested which provides excellent yield of the desired products (Table 3, entry 5 and 6). However, in case of strong electron-withdrawing groups (–NO2) yields of resultant products decreased (Table 3, entry 7). In case of other substituents such as 4-CN and 3-F the furnished 95% and 82% yields of respective amides (Table 3, entry 8 and 9). Unfortunately, –OH and –NH2 substituted aryl iodides were too sluggish to provide corresponding amides (results are not shown in table). Interestingly, heteroaromatic iodides including 3-iodopyridine, 2-iodopyridine and 2-iodothiophene remarkably work under the given conditions and afforded good yields of the corresponding amides (Table 3, entries 10–12).

























The recyclability of catalyst reduces the cost of process chemistry makes the protocol economically valuable and eliminates the rescale process. In this context, the recyclability study of Pd/C was further explored for synthesis of primary aromatic amides by carbonylation of 1a with ammonium carbamate under the optimized reaction conditions. After completion of the reaction the reactor was cooled to room temperature and remaining CO was carefully vented off. The catalyst was recovered by simple filtration from reaction mixtures and then washed three times with distilled water and finally by methanol to remove traces of organic contents.
After, washing and drying resulting catalyst was recycled up to four consecutive run (Fig. 2). A good catalytic activity was observed up to the 4th reaction run. The reaction mixtures after catalyst separation at the end of each cycle were subjected to ICP-AES analysis. No detectable amounts of palladium (below 0.01 ppm) were present in these samples indicating a negligible or no catalyst leaching.
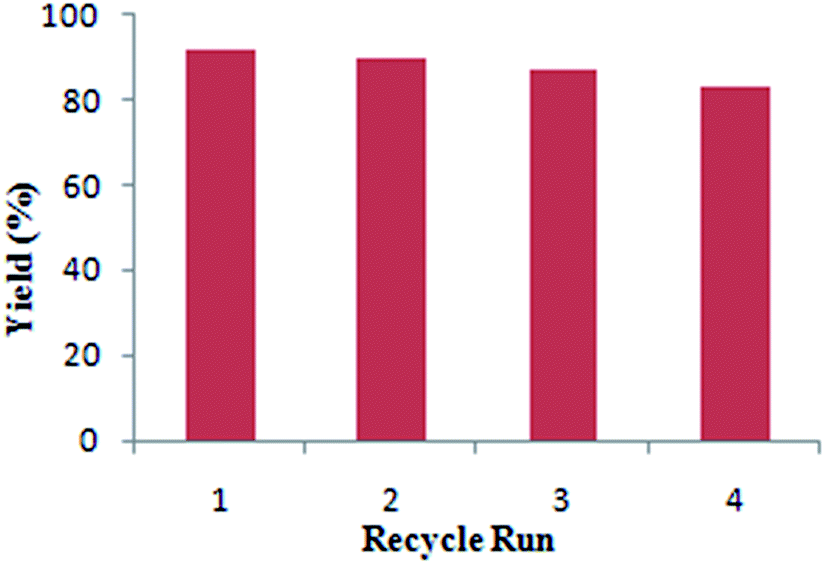 | ||
| Fig. 2 Recyclability study of Pd/C Catalyst. Reaction conditions: 1a (1 mmol), ammonium carbamate (1.5 mmol), 2.5 mol% Pd/C, KI (0.5 mmol), DABCO (2 mmol), MeCN (10 mL), CO (10 atm), 8 h, 90 °C. Yield determined by GC. | ||
A possible reaction mechanism for the aminocarbonylation has been illustrated (Scheme 2).10,21 The additive (KI) could play dual role i.e., it stabilises Pd/C catalyst or may acts as ligand in the absences of phosphine to prevent the deactivation of catalyst.23 And, then could be results into the formation of in situ active anionic Pd–I− species (I). The iodide can be adsorbs and desorbs easily from the catalyst surface as the iodide has ‘softer’ binding nature. First, reaction proceeds through the oxidative addition of aryl iodide 1 to the active anionic Pd–I− species to form complex (II). Subsequently, insertion of CO to complex (II) produced an acylpalladium complex (III). Then nucleophilic attack of ammonia to complex (III), which is in situ released from ammonium carbamate under the mild heating to form (IV). Followed by reductive elimination to form desired product 3 and regenerate the active catalytic species (I).
 | ||
| Scheme 2 Reaction mechanism for carbonylative synthesis of primary amides. | ||
Conclusion
In conclusion, we have developed a simple and efficient protocol for the synthesis of primary amides by aminocarbonylation of aryl iodides. The present methodology demonstrates a simple solid ammonia source instead of corrosive, hazardous and difficult to handle ammonia gas. The present system eliminates the use of air/moisture sensitive, expensive phosphine ligands and demonstrates development of the heterogeneous and recyclable catalytic system using commercially available Pd/C catalyst. The developed methodology tolerates wide range of electron donating and electron withdrawing functional groups and afforded the respective primary amides in moderate to excellent yields. The catalyst is recovered by simple filtration and effectively recycled to four consecutive run without loss in the catalytic activity.Experimental section
General
The Pd/C was purchased from Sigma-Aldrich (10 wt% loading, matrix: activated carbon support, product number: 205699, brand: Aldrich). All chemicals were purchased from Sigma Aldrich, Alfa Aesar and commercial suppliers. The resulting products were purified by column chromatography on silica gel (100–200 mesh; petroleum ether/ethyl acetate). All the products were confirmed by GC (Perkin Elmer Clarus 400) instrument with FID detector and capillary column (Elite – 1, 30 m, 0.32 mm ID, 0.25 μm film thickness) using N2 as carrier gas. GC-MS (Shimadzu QP 2010) instrument with EI detector and capillary column (Elite – 1, 30 m, 0.32 mm ID, 0.25 μm film thickness) using helium carrier gas. 1H and 13C NMR spectra were recorded with 400 MHz, 200 MHz and 100 MHz spectrometer respectively and compared with those of authentic data.General experimental procedure for carbonylative synthesis of primary aromatic amides
To a 100 mL stainless steel autoclave containing, aryl iodide (1 mmol), ammonium carbamate (1.5 mmol), Pd/C (2.5 mol%), KI (0.5 mmol), and DABCO (2 mmol) in a 10 mL acetonitrile were transferred under normal atmosphere. The reactor was closed, flushed with CO then pressurized with 10 bar of carbon monoxide, heated to 90 °C with constant stirring for 8 h. After the completion of reaction, the reactor cooled down to room temperature and remaining CO gas was carefully vented. The resultant reaction mixture filtered off by simple filtration. The filtrate was then quenched in water and the product was extracted with ethyl acetate (3 × 15 mL). The organic layer was then washed with brine and dried over Na2SO4 and concentrated on reduced pressure. The crude product was purified by column chromatography on silica gel (100–200 mesh; petroleum ether/ethyl acetate) and analyzed by GC and GC-MS. The products were confirmed by comparison of their GC-MS, 1H and 13C NMR spectroscopy with those of authentic data.Characterization of products
Acknowledgements
The author R. S. Mane gratefully acknowledges the Council of Scientific & Industrial Research (CSIR), India for providing Junior Research Fellowship (JRF).Notes and references
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed; (b) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS PubMed; (c) K. Badgujar, K. Dhake and B. M. Bhanage, Process Biochem., 2013, 48, 1335 CrossRef CAS PubMed.
- (a) C. E. Mabermann, in Encyclopedia of Chemical Technology, ed. J. I.Kroschwitz, Wiley, New York, 1991, vol. 1, p. 251 Search PubMed; (b) D. Lipp, in Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, Wiley, New York, 1991, vol. 1, p. 266 Search PubMed; (c) R. Opsahl, in Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, Wiley, New York, 1991, vol. 2, p. 346 Search PubMed.
- (a) K. Soai, A. Ookawa and H. Hayashi, J. Chem. Soc., Chem. Commun., 1983, 668 RSC; (b) A. Prasad, J. Kanth and M. Periasamy, Tetrahedron, 1992, 48, 4623 CrossRef CAS.
- (a) D. Bose and B. Jayalakshmi, J. Org. Chem., 1999, 64, 1713 CrossRef CAS; (b) K. Chaudhari, U. Mahajan, D. Bhalerao and K. Akamanchi, Synlett, 2007, 18, 2815 Search PubMed.
- (a) F. L. Becerra, P. A. Ojeda and S. D. Gamba, J. Org. Chem., 2014, 79, 4544 CrossRef PubMed; (b) S. N. Rao, D. C. Mohan and S. Adimurthy, Green Chem., 2014, 16, 4122 RSC.
- (a) T. Reginald, E. D. Steven, N. Noreen and M. Frank, J. Chem. Soc., Chem. Commun., 1978, 562 Search PubMed; (b) K. Tanaka, S. Yoshifuji and Y. Nitta, Chem. Pharm. Bull., 1988, 36, 3125 CrossRef CAS.
- (a) N. A. Owston, A. J. Parkar and J. M. J. Williams, Org. Lett., 2007, 9, 73 CrossRef CAS PubMed; (b) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC.
- (a) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC; (b) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Jr Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC; (c) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS PubMed.
- (a) K. N. Ali, P. Abolfath, N. S. R. Mohammad and Z. Abdolkarim, Tetrahedron Lett., 2005, 46, 6879 CrossRef PubMed; (b) J. C. Sheehan and G. P. Hess, J. Am. Chem. Soc., 1955, 77, 1067 CrossRef CAS; (c) T. Yasuhara, Y. Nagaka and K. J. Tomioka, J. Chem. Soc., Perkin Trans. 1, 2000, 1, 2901 RSC; (d) V. G. Jadhav, J. M. Bhojane and J. M. Nagarkar, RSC Adv., 2015, 5, 6636 RSC.
- (a) S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867 CrossRef CAS PubMed; (b) M. V. Khedkar, T. Sasaki and B. M. Bhanage, ACS Catal., 2013, 3, 287 CrossRef CAS; (c) A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4133 CrossRef PubMed.
- (a) T. M. Konrad, J. A. Fuentes, A. M. Z. Slawin and M. L. Clarke, Angew. Chem., Int. Ed., 2010, 49, 9197 CrossRef CAS PubMed; (b) D. B. G. Williams, M. L. Shaw, M. J. Green and C. W. Holzapfel, Angew. Chem., Int. Ed., 2008, 47, 560 CrossRef CAS PubMed; (c) T. O. Vieira, M. J. Green and H. Alper, Org. Lett., 2006, 8, 6143 CrossRef CAS PubMed; (d) W. Kim, K. Park, A. Park, J. Choe and S. Lee, Org. Lett., 2013, 15, 1654 CrossRef CAS PubMed; (e) T. Kippo, K. Hamaoka and I. Ryu, J. Am. Chem. Soc., 2013, 135, 632 CrossRef CAS PubMed; (f) K. Takahashi, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 18746 CrossRef CAS PubMed; (g) R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686 CrossRef CAS PubMed; (h) S. T. Gadge and B. M. Bhanage, RSC Adv., 2014, 4, 10367 RSC; (i) I. Fleischer, K. M. Dyballa, R. Jennerjahn, R. Jackstell, R. Franke, A. Spannenberg and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2949 CrossRef CAS PubMed; (j) J. Ferguson, F. Zeng and H. Alper, Org. Lett., 2012, 14, 5602 CrossRef CAS PubMed; (k) M. V. Khedkar, T. Sasaki and B. M. Bhanage, RSC Adv., 2013, 3, 7791 RSC.
- (a) X.-F. Wu, H. Neumann and M. Beller, Chem.–Asian J., 2010, 5, 2168 CrossRef CAS PubMed; (b) X.-F. Wu, J. Schranck, H. Neumann and M. Beller, ChemCatChem, 2012, 4, 69 CrossRef CAS PubMed; (c) G. Alsabeh, M. Stradiotto, H. Neumann and M. Beller, Adv. Synth. Catal., 2012, 354, 3065 CrossRef PubMed.
- E. Morera and G. Ortar, Tetrahedron Lett., 1998, 39, 2835 CrossRef CAS.
- (a) A. Schnyder, M. Beller, G. Mehltretter, T. Nsenda, M. Studer and A. F. Indolese, J. Org. Chem., 2001, 66, 4311 CrossRef CAS PubMed; (b) A. Schnyder and A. F. Indolese, J. Org. Chem., 2002, 67, 594 CrossRef CAS PubMed; (c) Y. Wan, M. Alterman, M. Larhed and A. Hallberg, J. Comb. Chem., 2003, 5, 82 CrossRef CAS PubMed.
- T. Eszter, V. Csilla, R. Skoda-Foldes and L. Kollar, Tetrahedron Lett., 2007, 48, 2453 CrossRef PubMed.
- X. Wu, J. Wannberg and M. Larhed, Tetrahedron, 2006, 62, 4665 CrossRef CAS PubMed.
- K. Ueda, Y. Sato and M. Mori, J. Am. Chem. Soc., 2000, 122, 10722 CrossRef CAS.
- (a) D. U. Nielsen, R. H. Taaning, A. T. Lindhardt, T. M. Gogsig and T. Skrydstrup, Org. Lett., 2011, 13, 4454 CrossRef CAS PubMed; (b) J. Balogh, S. Maho, V. Hada, L. Kollar and R. Skoda-Foldes, Synthesis, 2008, 3040 CAS.
- T. Xu and H. Alper, Tetrahedron Lett., 2013, 54, 5496 CrossRef CAS PubMed.
- S. T. Gadge and B. M. Bhanage, Synlett, 2014, 25, 85 CAS.
- (a) P. J. Tambade, Y. P. Patil, M. J. Bhanushali and B. M. Bhanage, Tetrahedron Lett., 2008, 49, 2221 CrossRef CAS PubMed; (b) M. V. Khedkar, S. R. Khan, D. N. Sawant, D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2011, 353, 3415 CrossRef CAS PubMed; (c) M. V. Khedkar, P. J. Tambade, Z. S. Qureshi and B. M. Bhanage, Eur. J. Org. Chem., 2010, 6981 CrossRef CAS PubMed; (d) S. T. Gadge, M. V. Khedkar, S. R. Lanke and B. M. Bhanage, Adv. Synth. Catal., 2012, 354, 1 CrossRef PubMed; (e) S. P. Chavan and B. M. Bhanage, Tetrahedron Lett., 2014, 55, 1199 CrossRef CAS PubMed; (f) J. Liu, J. Chen and C. Xia, J. Catal., 2008, 253, 50 CrossRef CAS PubMed.
- F. Tinnis, H. Lundberg and H. Adolfssona, Adv. Synth. Catal., 2012, 354, 2531 CrossRef CAS PubMed.
- (a) J. G. de Vries, Dalton Trans., 2006, 421, 421 RSC; (b) C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314 CrossRef CAS PubMed; (c) S. P. Gupte and R. V. Chaudhari, J. Catal., 1988, 114, 246 CrossRef CAS; (d) F. Zhao, B. M. Bhanage, M. Shirai and M. Arai, Chem.–Eur. J., 2000, 6, 843 CrossRef CAS.
- S. C. Ghosh, J. S. Ngiam and A. M. Seayad, J. Org. Chem., 2012, 77, 8007 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, experimental procedure, 1H, 13C NMR and GC-MS data of products. See DOI: 10.1039/c5ra15831a |
| This journal is © The Royal Society of Chemistry 2015 |