
The enhancement of liquid–liquid extraction with high phase ratio by microfluidic-based hollow droplet†
Wen-Ting
Wang
,
Fu-Ning
Sang
,
Jian-Hong
Xu
*,
Yun-Dong
Wang
and
Guang-Sheng
Luo
The State Key Lab of Chemical Engineering, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: xujianhong@tsinghua.edu.cn; Fax: +86-10-62773017; Tel: +86-10-62773017
First published on 9th September 2015
Abstract
In this study, a one-step microfluidic capillary device for the enhancement of mass transfer process with high phase ratio by hollow droplet was developed. Herein, a fluorescent material rhodamine B was used as the transportation substance, which gets transferred from an aqueous phase to an oil phase. Qualitative demonstrations of the extraction process are presented. During the droplet formation stage, the fluorescent brightness of the droplets becomes stronger for a droplet with and without a bubble in it. During the droplet moving stage, for both flow conditions, the fluorescence intensity increases along the outlet channel. At the latter half of the outlet channel, the addition of gas greatly enhances the mass transfer process. The mean overall volumetric mass transfer coefficients kLa increases with increasing gas flow rate; this is mostly because of the sharply increased specific area. Moreover, the kLa value of gas–liquid–liquid hollow droplet flow increases around 10–60 times compared to that of a liquid–liquid droplet flow system. The length of extraction equipment needed to reach 95% extraction efficiency is reduced to around 10–1000 times when gas microbubbles are introduced. Based on the experimental data, a theoretical model has been built up for the potential prediction of the enhancement of the extraction by adding gas microbubbles. The effective diffusion coefficient is introduced to combine the convective mass transfer factor into this model. The modelling results fit well with the experimental data. All the abovementioned results present a practical method for the enhancement of the extraction process with high phase ratio systems, which has potential applications in analytical chemistry, micro-extraction, and biological extraction.
Introduction
Liquid–liquid extraction technology is an important separation unit and it is widely applied in many fields such as chemical engineering, biology, material fabrication and analytical chemistry. However, in chemical industry, traditional extraction equipment has limited extraction capability with particular extraction systems. This always leads to large-scale volumes of equipment and a very high energy cost, as is experienced in chemical factories. The development of novel extraction equipment is very important for the chemical industry and environmental protection.Recently, liquid–liquid extraction based on microfluidic technology has been developed rapidly not only to reduce the sample and increase the portability, but also for the better mass transfer performance than the conventional equipment such as extraction columns and mixer-settlers. For example, Burns et al.1 developed a multiphase micro reactor based upon the use of slug flow through a narrow channel. The internal circulation was found to be the main reason for a large enhancement in the interfacial mass transfer from the aqueous phase to the oil phase. The experimental data showed a better extraction performance with microfluidic-based slug flow than conventional extraction methods. Kashid et al.2 investigated the mass transfer performance of slug-flow for various operating conditions in a capillary microstructured reactor. Compared to conventional contactors, the mass transfer coefficients were approximately one order of magnitude higher, allowing for process intensification. The results obtained demonstrate the benefits of microstructured reactors and confirm that the throughput of conventional reactors can be achieved. Pascaline et al.3 analysed the extraction performance of fluorescein and rhodamine in drop flow and the influences of different parameters on the process were discussed. Xu et al.4 studied the enhancement of the mass transfer performance by droplet flow in a co-flowing microchannel. The data has been obtained by monitoring the extraction of succinic acid from n-butanol to aqueous drops containing NaOH. The mass transfer rate was 10–1000 times higher compared to the traditional liquid–liquid systems. The results from this study indicate that droplet flow within a micrometer scale environment offers a viable alternative for a two-phase reaction or separation process. Benz et al.5 developed micro mixer arrays to evaluate the extraction performance of miniplant technology as an alternative to conventional stirring apparatuses. The study proved that micro mixer arrays are highly efficient apparatuses for extraction purposes. Despite the circulations in a particular flow pattern, such as slug flow, there are other reasons for the enhancement of extraction in micro devices. In micro mixers, the dispersed phase can form in considerably smaller droplets and thus they will have larger specific area and shorter mass transfer distance than in ordinary-size equipment, which leads to a faster mass transport rate. Thus, in microfluidic devices, it takes as little as 1 s to achieve a liquid–liquid extraction equilibrium, which means it is environmentally friendly and takes less energy in industry.
Based on these basic investigations on liquid–liquid extraction in microfluidic devices, this new method has been applied in the extraction of compounds of biological relevance,6–13 metal ions14–18 and other chemicals.19–24 Wagli et al.12 combined a microfluidic-based multiphase liquid–liquid extraction method to continuously extract cocaine from IR-light-absorbing saliva to an IR-transparent solvent. They succeeded in detecting cocaine in real saliva samples spiked with the drug and allowed real time measurements, which makes this approach suitable for point-of-care applications. Nam et al.11 described the separation of live and dead cells of Chinese hamster ovary cells by a change of overall surface properties of animal cells such as hydrophobicity and surface net charge. In their study, all live cells were partitioned to PEG-phase flow at the junction of outlet microchannel, while most of the dead cells were kept on the inter-face flow at pH 6.6. Maruyama et al.18 studied a three-phase flow in a microchannel on a glass microchip. They demonstrated that the conventional methodology for solvent extraction of metal ions is applicable to solvent extraction in a microchannel. They succeeded in the selective separation of a targeted metal ion from an aqueous feed solution to a receiving phase within a few seconds by employing a liquid membrane formed within a microfluidic device.
However, for extraction processes with a high phase ratio (usually higher than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which is far away from 1:1, such as oil refining processes,25 caprolactam extraction,26 phosphoric acid purification,27,28 hydrogen peroxide extraction,29 and most of the micro-extraction processes,30 the mass transfer distance in the continuous phase will be larger and the contact area between the two phases will decrease sharply as the phase ratio increases. Therefore, external energy input is greatly needed for these high phase ratio extraction processes. The development of a more efficient extraction method and equipment is needed to overcome the abovementioned difficulties caused by the high phase ratio. Tan et al.31 designed a gas-agitated method for the intensification of H2O2 extraction with a high phase ratio, which is larger than 50:1. A device with microfiltration membranes as the dispersion medium was developed to generate the gas–liquid–liquid micro dispersed system. The research results show that the overall volume mass transfer coefficient could be 10–30 times larger than the extraction process without gas addition. However, the detail structure of gas–liquid–liquid system during the extraction process has not been presented, and the mechanism of the enhancement of extraction by adding gas has not been systematically analysed.
:1), which is far away from 1:1, such as oil refining processes,25 caprolactam extraction,26 phosphoric acid purification,27,28 hydrogen peroxide extraction,29 and most of the micro-extraction processes,30 the mass transfer distance in the continuous phase will be larger and the contact area between the two phases will decrease sharply as the phase ratio increases. Therefore, external energy input is greatly needed for these high phase ratio extraction processes. The development of a more efficient extraction method and equipment is needed to overcome the abovementioned difficulties caused by the high phase ratio. Tan et al.31 designed a gas-agitated method for the intensification of H2O2 extraction with a high phase ratio, which is larger than 50:1. A device with microfiltration membranes as the dispersion medium was developed to generate the gas–liquid–liquid micro dispersed system. The research results show that the overall volume mass transfer coefficient could be 10–30 times larger than the extraction process without gas addition. However, the detail structure of gas–liquid–liquid system during the extraction process has not been presented, and the mechanism of the enhancement of extraction by adding gas has not been systematically analysed.
In this study, we used a simple one-step microfluidic device for the formation of gas in oil in a water dispersed system (also known as a hollow droplet structure) to realize the enhancement of the extraction process from the aqueous phase to the oil phase with a high phase ratio. A stable gas-in-oil-in-water hollow droplet structure (usually called double emulsions) can be easily fabricated with this microfluidic device. In our previous studies,32–35 we had systematically analysed the generation of gas-in-liquid-in-liquid double emulsions and the structural control by changing the system properties and operating conditions, as well as the formation mechanism of these types of double emulsions. These previous studies have made a good foundation for this study that concerns the enhancement of extraction behaviour of the hollow droplet structure. Herein, we used a fluorescent material rhodamine B as the transportation substance, which transfers from the aqueous phase to organic phase due to the contrast in its chemical potential between the two phases. The qualitative and quantitative demonstrations of the enhancement of the extraction process have been presented. The effects of equipment length, gas phase flow rates and phase ratios on the extraction process have been systematically studied and analysed. The mass transfer coefficients when extraction efficiency is 95% are calculated and discussed. Based on the experimental data, a theoretical model has been built up for the prediction of the enhancement of extraction by this type of gas-filled hollow droplet.
Experimental
Microfluidic device
The experimental set-up is shown in Fig. 1. The most important part of this device is the dual-coaxial capillary structure, where the oil droplet with a gas bubble inside it (gas-in-oil-in-water hollow droplet structure) is formed by the shearing force of continuous aqueous phase flow (ESI MOV1†). To fabricate this device, a smaller capillary is injected into another capillary and the tapered orifices are aligned where the one-step emulsification occurs; the image shown in the rectangle with green dotted sides in Fig. 1 demonstrates this process. A circular glass capillary with an inner-diameter of 0.70 mm and an outer-diameter of 1.00 mm was tapered using a micropipet puller (P-97, SUTTER Co. Ltd, USA) for the injection of the gas phase fluid. The diameter of the tapered orifice is approximately 20 μm. The first tapered capillary was inserted into another capillary with an inner-diameter of 1.05 mm and an outer-diameter of 1.50 mm for the middle phase and the orifice was tapered to approximately 250 μm. We made sure that both orifices aligned in the same plane. Then, these compound capillaries were inserted into the outlet channel, a third coaxially aligned capillary. The inner-diameter of the third capillary was 1.05 mm and the outer-diameter was 1.5 mm. We changed the length of the outlet channel to control the residence time of the two liquid phases. The capillaries were fixed on a cross-junction channel, which was fabricated on a polymethyl methacrylate (PMMA) plate using a computerized numerical control (CNC) machine tool with an end mill (Φ = 1.0 mm). The channels for the dispersed phase fluid and the continuous phase fluid are both approximately 1.5 mm wide × 1.5 mm high. PTFE pipes were inserted into the inlet channel and micro syringe pumps (LSP01-1B, Baoding Longer Precision Pump Co., Ltd) were used to pump the gas phase and two liquid phases into the microfluidic device. The outlet channel, the phase separator made of a silicon tube with ∅ = 6 mm × 7 mm, h = 8 cm, and the exit made by a glass capillary with an inner-diameter of 1.05 mm made a T union, which was fixed on a T-junction channel fabricated on another PMMA plate. All the three phases flowed through the outlet channel, into the separator. The phase separation was achieved by density difference of the three phases. The phases with low density, namely, oil phase and gas phase, floated up to the top surface of the separator. The oil phase accumulated on the top surface and was pumped out into the sample reservoir. The gas phase escaped into the air atmosphere as soon as it came into contact with the atmosphere. Because gas is considerably lighter than liquid, the addition of gas can intensify the phase separation process. The high-density continuous aqueous phase, however, flowed past the separator, straight to the exit, which was at the bottom of the separator. The aqueous phase could then be collected in the sample reservoir.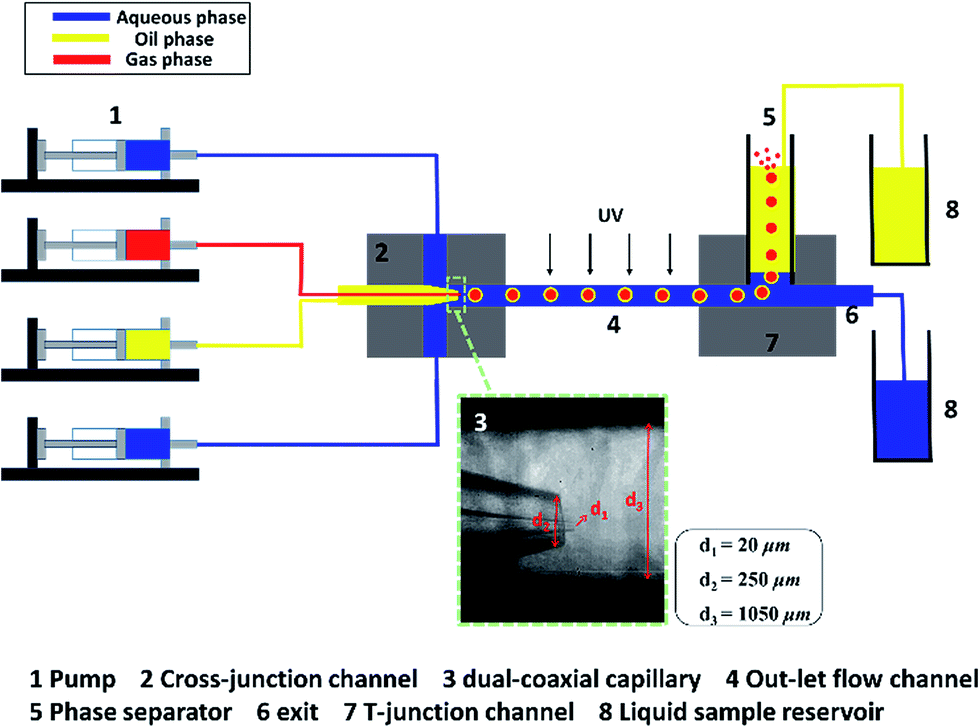 | ||
| Fig. 1 The structure of the microfluidic device. | ||
The experimental process was operated under a fluorescent microscope (DSU, Olympus) with an UV irradiation of 450–650 nm as the exciting light of rhodamine B, and the flow condition and the fluorescence intensity of the outlet flow was observed by a high-speed video camera (Phantom 663, 2500 fps) mounted on the microscope.
Materials
In our experiments, air gas, as the inner phase, breaks up into microbubbles at the orifice, and gets dispersed into the oil phase (49 wt% silicon oil (10 cSt) + 49 wt% octanol + 2 wt% Dowcorning 749). Then, the oil droplets encapsulating a gas bubble each were sheared off by the outer aqueous phase flow, which contains 2.5 wt% polyethylene glycol and rhodamine B (95%, J&K) in concentrations of 10.0 mg L−1. The distribution coefficient of rhodamine B between the oil and aqueous phase is 46, which is obtained from our extraction experimental data with shake flasks. The viscosity of the aqueous phase is 2.60 mPa s, which is measured by a rotor viscosity meter (LVDV-II+ PRO, Brookfild). The surface/interfacial tensions of the gas–aqueous, gas–oil, aqueous–oil are 50.46 mN m−1, 23.42 mN m−1, 7.85 mN m−1 respectively, which indicate that the gas-in-oil-in-water double emulsions will be a thermodynamically steady structure.36Analysis
Two types of experimental results have been obtained from the analysis, qualitative ones and quantitative ones. The qualitative results include the fluorescence intensity of the oil droplets, which can be observed from the fluorescent microscope. As rhodamine B transported from the aqueous phase to the organic phase, the concentration of rhodamine B in oil phase increases along the outlet flow channel. According to the Beer–Lambert law, the fluorescence intensity is linear to the concentration of rhodamine B, when it is at low concentrations. Therefore, the fluorescence intensity increases along the outlet flow channel.The quantitative results are obtained by measuring concentrations of the aqueous phase samples collected after the phase separation reservoir. A UV-visible spectrophotometer (UV-2450, Shimadzu) was used to measure the concentration of rhodamine B in the aqueous phase. According to the spectrum (Fig. S1 in ESI†), the maximum absorption wavelength of rhodamine B is 550 nm. The standard curve (Fig. S2 in ESI†) shows that the absorbance is linear with concentration when it is below 20 mg L−1.
Theoretical background
Mass transfer equation
In the extraction process, the mass transfer resistances can be presented in the form of eqn (1):37,38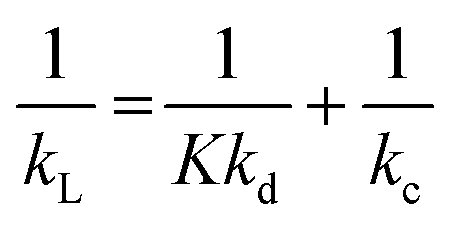 | (1) |
 are the mass transfer resistances of the overall system, dispersed phase and continuous phase, respectively. K is the distribution coefficient of the dispersed phase and continuous phase.
are the mass transfer resistances of the overall system, dispersed phase and continuous phase, respectively. K is the distribution coefficient of the dispersed phase and continuous phase.
When K ≪ 1, the mass transfer resistance in the dispersed phases is considerably larger than that of continuous phase; thus, the resistance in the continuous phase can be ignored. The overall mass transfer coefficient is kL = Kkd. When K ≫ 1, on the contrary, kL = kc. For our experimental system, K = 46 ≫ 1; thus, the overall mass transfer coefficient is kL = kc. In other words, in our experiments, the resistance in dispersed phase is ignored and we consider that the concentration in the dispersed phase becomes uniform instantly.
Mass transfer model for the continuous phase fluid
From the abovementioned analysis, only mass transfer performance outside the droplets is taken into account in our extraction system. The Sherwood number (Sh = kd/D), which is also called as the dimensionless mass transfer coefficient, is the ratio of the convective mass transfer to the molecular diffusion. For fully developed velocity and concentration profiles, Sh becomes constant and the mass transfer coefficient is inversely proportional to the characteristic length d. There are several models predicting Sh outside the droplets, which are summarized in different operating conditions in fully developed flow. The applicability of these models depends on the Peclet number (Pe = ud/D, in which u is the relative velocity of the continuous phase and a dispersed phase, m s−1) and Reynolds number (Re = ρdu/μ, ρ and μ are the density and viscosity of the continuous phase, kg m−3 and Pa s).For the condition39 where the continuous phase is still, Sh0 = 2. This is the minimum value of Sh outside the droplets and the applicable condition is Pe → 0.
For the conditions40 where the continuous flow pattern is creeping flow and Re ≤ 1, Pe ≫ 0:
| Shc = 1 + (1 + 0.564Pe2/3)3/4 | (2) |
For the conditions41 when Re > 1:
| Shc = 2.0 + 0.76Re1/2Sc1/3 | (3) |
Results and discussion
Qualitative analysis
Using observation with a fluorescent microscope, the mass transfer from the continuous aqueous phase to the oil droplets along the outlet channel could be qualitatively demonstrated. The extraction process can be divided into two steps: the droplet formation stage and droplet moving stage.Droplets formation stage
The extraction performance within the droplet formation stage is shown in Fig. 2a and b and MOV2, MOV3 in the ESI.† The aqueous and organic flow rates are 600 μL min−1 and 6 μL min−1, respectively, both for the two operating conditions, and for MOV3 there is additional gas flow with the flow rate of 16 μL min−1. We can see that for both operating conditions (MOV2, MOV3† and dotted outlines in Fig. 2a and b), it is obvious that, as the droplet grows at the tip of the capillary, the fluorescent brightness of the droplets becomes more and more strong. This is because as long as the two phases come into contact, rhodamine B starts getting transported from the continuous aqueous phase into the oil phase droplet. The brightness starts at the edges of the droplet, and then spreads inside the droplets. Because the distribution coefficient is a relatively high value (46), the brightness inside the dispersed liquid phase and at the edge of the phase interfaces could be considerably higher than in the continuous phase when the transformation occurs. Because the mass transfer between the two liquid phases occurs at the phase interface, the brightness of the interface should light up first and strongly as well. Thus, the edge of the droplet is so clear. In the dotted rectangle shown in Fig. 2a and b, the bright areas at the phase interfaces and within the droplets represent the location where rhodamine B molecules exist. The brighter the area is, the higher the rhodamine B concentration is. Thus, we can qualitatively infer the fluorescence intensity and the rhodamine B concentration by the brightness of the droplet. For the droplets without a bubble, we can also observe that the fluorescent brightness spreads along two incomplete arcs, which are symmetrical to the axis of the droplets (Fig. 2b). This phenomenon could be explained by the inner loop of the droplets during the formation process. This internal circulation is the main factor that increases the extraction rate. For the gas-in-oil hollow droplets, the generation frequency (f) is higher than simple droplets (Fig. 2c). During the formation stage, the oil phase first flows out of the annulus of the two capillary tips, and when the oil sphere grows to a certain size, the gas phase bubble comes out of the inner capillary tip and then the whole hollow droplet is sheared off by the outer phase flow. Because of the additional bubble, there is no internal circulation in the organic phase and the total fluorescence intensity is weaker than that of the simple oil droplet during the droplet formation stage. The gas phase exhibits no fluorescence response to the UV light; therefore, a black round feature could be observed when the gas bubble is introduced (Fig. 2a). | ||
| Fig. 2 (a) Fluorescence images during droplet formation stage with microbubble inside it. The dotted outlines highlight the droplets (Qa = 600 μL min−1, Qo = 6 μL min−1, Qg = 16 μL min−1). (b) Fluorescence images during droplet formation stage without a microbubble (Qa = 600 μL min−1, Qo = 6 μL min−1 and Qg = 0 μL min−1). (c) The generation droplet formation frequency with different gas phase to oil phase ratios. Qa = 600 μL min−1. (d) Fluorescence images of gas-in-oil-in-water flow conditions at different locations of the outlet channel during the droplet moving stage (Qa = 600 μL min−1, Qo = 6 μL min−1, Qg = 16 μL min−1). (e) Fluorescence images of liquid–liquid flow conditions at different locations of the outlet channel during the droplet moving stage (Qa = 600 μL min−1, Qo = 6 μL min−1 and Qg = 16 μL min−1). (f) The images on the left show the different generation frequencies when the inner gas flow rates are increased from 0 to a high value. In the middle is a sketch of the dispersion states inside the outlet channel before and after the gas microbubble is introduced. The images on the right show the increase of fluorescence intensity when the gas phase is added. | ||
Droplet moving stage
For the qualitative demonstration during the droplet moving stage in the outlet channel, MOV4 and MOV 5 in the ESI† show the videos of two flow conditions, which are captured at the same location, i.e. 10 cm away from the dispersing point. The aqueous and oil flow rates are 600 μL min−1 and 6 μL min−1, respectively, for both the conditions, and for MOV5, there is an additional gas flow with a flow rate of 16 μL min−1. Fig. 2d and e show the fluorescence images of two flow conditions at different locations of the outlet channel. To enhance the visual contrast, we treated the original images with a deduction of background grayscale. The image processing method is shown in the ESI.† The figures below the images represent the distance away from the droplet formation tip, and the flow rates are Qa = 600 μL min−1 and Qo = 6 μL min−1 for both conditions and Qg = 16 μL min−1 for Fig. 2d (condition A) and Qg = 0 μL min−1 for Fig. 2e (condition B). We can see that for both flow conditions, the fluorescence intensity increases along the outlet channel because of the transport of rhodamine B into droplets as long as the contact time of two phases lasts. At the location of 2 cm, the fluorescence intensity of condition B is higher than that of condition A; this is because this location is not far away from the droplet formation stage, after which the extraction performance of single oil droplets is better than that of gas-in-oil hollow droplets. However, as the flow continues, for example at the points from 6 to 8 cm, the fluorescence intensity of condition A becomes higher than condition B, which means the total extraction performance of condition A is better. For the first reason, the internal circulation inside the simple oil droplet disappears after the droplet breakup at the capillary tip, and the enhancement of mass transfer by the internal circulation disappears. For the second reason, as shown in Fig. 2f, the addition of a microbubble inside the oil droplet shortened the mass transfer distance in the oil phase because the oil droplet becomes a thin oil membrane that spreads on the surface of the microbubble. The mass transfer distance shortens from the radius of the droplet (around 350 μm) to the thickness of the thin membrane (around 20 μm). For the third reason, the mass transfer distance in the aqueous phase is shortened largely. When gas phase is introduced, the generation frequency increases; therefore, the space interval of every two droplets decreases, and thus most of the rhodamine B molecules can travel a shorter distance to reach the interfaces of the two phases. For the fourth reason, the contact area of two liquid phases also increases as the droplet becomes denser when the gas microbubble is added.Quantitative analysis
To obtain the specific extraction performance of the two flow conditions, the desired concentrations of rhodamine B during the extraction process must be obtained. The accurate data was obtained by analysing the concentrations of aqueous phase samples collected after the micro-phase separator, as shown in Fig. 1. We varied the length of outlet channel from 5 cm to 30 cm with intervals of 5 cm for different residence times. We define extraction efficiency E in eqn (4).| E = (C0 − Cext)/(C0 − C*) | (4) |
| QaC0 + Qo × 0 = QaC* + QoKC* | (5) |
:1, E increases from 40% to 95% with the increase of inner gas flow rate, which indicates that the mass transfer rate is highly increased. Fig. 3b–d show that when the aqueous to oil phase ratios are at a relatively lower value, 59:1, 40:1 and 20:1, E increases sharply as long as the gas phase is introduced. However, when the gas phase flow rate increases, E only tends to increase in a small range, which is within 10% in our operating conditions. This is probably because the different gas phase rate ranges among different aqueous to organic phase ratios. Because the oil phase flow rate increases, it tends to hold a relatively high gas flow rate to stabilize the flow. For example, the gas flow rate ranges from 180 μL min−1 to 500 μL min−1 for Fig. 3a, and it ranges from 250 μL min−1 to 600 μL min−1, from 250 μL min−1 to 700 μL min−1 and from 700 μL min−1 to 1400 μL min−1 for Fig. 3b–d, respectively. As shown in eqn (6), when Qg is high enough to match Qa, the higher gas phase ratio easily makes a faster droplet moving velocity, which leads to a shorter residence time at the same outlet channel length. As the residence time decreases, E cannot increase obviously, even though more gas is introduced.| u = (Qa + Qo + Qg)/A | (6) |
 | ||
| Fig. 3 (a–d) The influences of Qg and L on the extraction efficiency when Qo = 10, 17, 25, and 50 μL min−1, respectively. The RSDs are less than 5%. (e) The influence of gas to oil phase ratio Qg/Qo and oil phase flow rate Qo on kLa. (f) The specific area variation as the increase of Qg/Qo (Qo = 17 μL min−1 and 50 μL min−1). (g–i) The variation of L(95%), kLa and kL, respectively, when E = 95% in different operating conditions. All the abovementioned data are obtained under the condition of Qa = 1000 μL min−1. | ||
The mean overall volumetric mass transfer coefficients (kLa, s−1) are usually used to represent the mass transfer characteristics in microchannels. kLa can be calculated by the following equations:
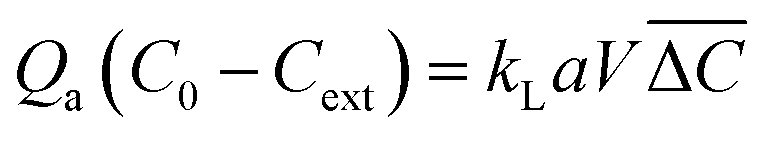 | (7) |
| a = 6φ/ddroplet | (8) |
| φ = (Qg + Qo)/(Qg + Qo + Qa) | (9) |
 | (10) |
| C*0 = Cm0/K, C*ext = Cmext/K | (11) |
| QaC0 + Qo × 0 = QaCext + QoC0ext | (12) |
 is the logarithmic mean concentration driving force, mg L−1C*0 is the equilibrium concentration of the solute in the inlet aqueous phase, corresponding to the actual inlet concentration of the solutes in the organic phase Cm0, which is 0. C*ext is the equilibrium concentration of the solutes in the outlet aqueous phase, corresponding to the actual outlet concentration of the solutes in the organic phase Cmext, which can be calculated by the mass conservation using eqn (12).
is the logarithmic mean concentration driving force, mg L−1C*0 is the equilibrium concentration of the solute in the inlet aqueous phase, corresponding to the actual inlet concentration of the solutes in the organic phase Cm0, which is 0. C*ext is the equilibrium concentration of the solutes in the outlet aqueous phase, corresponding to the actual outlet concentration of the solutes in the organic phase Cmext, which can be calculated by the mass conservation using eqn (12).
Herein, we choose the 30 cm length microchannel as the overall volume. Fig. 3e shows that the mean overall volumetric mass transfer coefficients kLa increases with the increase of gas to oil phase ratio. When Qg/Qo increases from 0 to 50, kLa becomes 2 to 10 times larger than that without a gas phase. This is mainly because of the sharp increase of specific area when the gas microbubbles are introduced, which is demonstrated in Fig. 3f.
To compare the extraction performances when the flow and mass transfer are well developed, we discussed the mean mass transfer coefficient kLa, which is obtained during the length interval, [0, L95%]. L95% is the channel length needed for E to reach 95%. Eqn (7) to (12) with some detailed correction can be applied to this calculation, where C95% replaces Cext and AL95% replaces V. C95% is the concentration in the aqueous phase when E = 95%, which can be calculated by eqn (4) and (5). A is the cross-sectional area of the outlet channel, m2. L95% is a value obtained by an experimental data fitted equation when C becomes C95%. Herein, an exponential function3C = M + Nexp(−PL) is applied to fit the experimental data. M, N and P are undetermined constants, and M and N are related to the concentrations of the inlet and outlet aqueous flow. The data of C and L in the range of 0–30 cm were used to fit the exponential function in different operating conditions.
Fig. 3g–i show the results of L95%, kLa and kL. kL is calculated by the equation kL = kLa/a. From Fig. 3g, we can see that the length of the extraction equipment needed to reach 95% extraction efficiency reduced around 10–1000 times when gas microbubbles are introduced. This means that when a specific extraction efficiency is required, the addition of gas can largely reduce the equipment size and even shorten the extraction time, not only because of the shorter length of the outlet channel but also due to the higher move velocity. As shown in Fig. 3h, kLa of gas–liquid–liquid hollow droplet flow increases around 10–60 times compared to that of liquid–liquid droplet flow system. The sharply increased specific area is the major contribution to this enhancement of extraction performance. The mass transfer coefficient kL is also a contribution, as shown in Fig. 3i. kL increases around 2–3 times when a gas phase is added. This phenomenon can roughly be explained by the two-film theory model,38 where kL = D/δ is presented. When the gas phase is introduced, the interval between two droplets decreases, which leads to the decrease of concentration boundary layer δ and increase of kL.
Theoretical model
To further analyse the mechanism of enhancement of the extraction process with the introduction of gas microbubbles, a theoretical model was built. Due to the steady flow condition, the mass transfer process in the outlet channel is divided into quantities of the same separate units (as shown in Fig. 4, inside red dotted rectangle). Each unit of gas–liquid–liquid fluid can be simplified into a sphere, which contains a gas bubble in the centre and two liquid films orderly spreading outside the bubble. The oil film is at the middle of the gas bubble and the aqueous film (Fig. 4a). Each unit of liquid–liquid flow can be simplified into a sphere, which contains an oil droplet in the centre and an aqueous film spreading on it. In the extraction process, the rhodamine B molecule transfers from the aqueous phase to the organic phase. | ||
| Fig. 4 Schematic representation of the mass transfer unit in G/O/W system and O/W system. | ||
The following assumptions are made for the modelling of the mass transfer process.
(1) The process is based on a diffusion model. The effective diffusion coefficient is used to combine the convective mass transfer factor into this model. The effective diffusion coefficients can be calculated by eqn (13):
| Deff = KfD | (13) |
| Kf = Shc/Sh | (14) |
As stated in the theoretical background part, Sh0 = 2. The formula of Shc is depended on Re. Fig. 5a shows the Re values in different operating conditions. For liquid–liquid flow, Re < 1, and eqn (2) is applied to calculate Shc. However, for gas–liquid–liquid flow, Re > 1, thus eqn (3) is applied. The effective diffusion coefficient is presented in Fig. 5b. Deff for liquid–liquid flow ranges from 6 × 10−9 m2 s−1 to 7 × 10−9 m2 s−1, and that for gas–liquid–liquid flow ranges from 8 × 10−9 m2 s−1 to 1 × 10−8 m2 s−1. Kf values ranges from 30 to 60.
 | ||
| Fig. 5 (a) The effect of Qg/Qo on Re. (b) The effect of Qg/Qo on Deff. (c and d) The comparison of extraction efficiency between experiment values and modelling results for Qa = 1000 μL min−1, Qo = 25 μL min−1 and Qa = 1000 μL min−1, Qo = 10 μL min−1, respectively. (e and f) The prediction of extraction efficiency on operating conditions of Qa = 1000 μL min−1, Qo = 25 μL min−1 and Qa = 1000 μL min−1, Qo = 17 μL min−1, respectively. | ||
(2) The overall radius R2 can be calculated by the droplet radius R1 as well as the flow rate ratio (eqn (15)). R1 can be measured with the captured images in the experiments.
 | (15) |
(3) The mass transfer resistance in oil phase is ignored according to eqn (1) in the section of theoretical background. In addition, the thickness of oil film is considerably small compared to the aqueous film. Therefore, the concentration in the organic phase is considered to be uniform.
(4) The mass transfer resistance at the aqueous–oil interface is ignored and the concentration at the interface is consistent with distribution coefficient.
(5) There is no mass transfer between every two neighbouring units because they are totally equivalent.
(6) Spherical coordinate is used and there is only mass transfer in the direction of r but no mass transfer in the other two directions.
With all the abovementioned assumptions, mass transfer through the aqueous film can be approximately described using the continuum model in the spherical coordinate, which is written as follows:
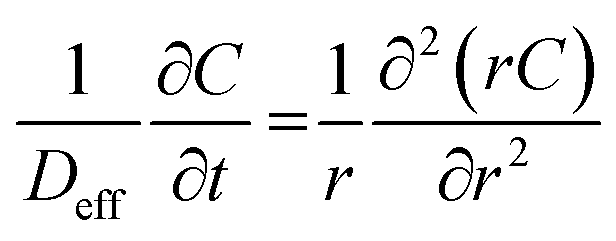 | (16) |
Initial condition: t = 0, R1 ≤ r ≤ R2, C = C0
Boundary conditions: 
| r = R1, C = Cit = Cm/K | (17) |
C 0 is the initial concentration in the aqueous phase.
C m is the concentration in organic phase according t, which can be calculated by mass conservation equation:
Qa × C0 = Qa × ![[C with combining macron]](https://www.rsc.org/images/entities/i_char_0043_0304.gif) + Qo × Cm + Qo × Cm | (18) |
is the average concentration in aqueous phase according to following equation:
 | (19) |
In the abovementioned model, C0 and R1 are given. The abovementioned model was analysed with numerical solutions by Matlab. Fig. 5c and d show the modelling results of extraction efficiency calculated by eqn (4), as well as the comparison with the experimental data. The length L = u × t and u is given by eqn (6). The modelling results show a good alignment with the experimental data. It is feasible to predict the extraction performance using this model. Fig. 5e and f are the results of prediction where the droplet size R1 is assumed to be 1 mm at all the operating conditions. Fig. 5e and f show that with a high aqueous to organic phase ratio, the increase of gas to organic phase ratio leads to an increase of extraction efficiency in the same equipment volume. When the gas to oil phase ratio is higher than 100, it takes less than a 10 cm length of equipment to complete the extraction. This prediction may not fit well with some of the real results, because some operating limits are not taken into consideration such as the coalescence of microbubbles and the instability of the flow condition when the gas to oil phase ratio is very high. However, it is still a practical model to obtain the approximate results within particular operating conditions.
Conclusions
In this study, we used a one-step microfluidic capillary device to obtain a gas-in-oil-in-water dispersed system (also known as a hollow droplet structure) to realize the enhancement of mass transfer with a high phase ratio by hollow droplet. Herein, we used a fluorescent material rhodamine B as the transportation substance, which transfers from an aqueous phase to an oil phase. The qualitative demonstrations of the extraction process have been presented. During the droplet formation stage, the fluorescent brightness of the droplets became stronger for the droplet. The inner convection inside the oil droplet was observed. During the droplet moving stage, for both flow conditions, the fluorescence intensity increased along the outlet channel, which is because more and more rhodamine B transports into droplets as long as the contact time of two phases lasts. At the latter half of the outlet channel, the fluorescence intensity of a droplet with a microbubble inside it became higher than a single droplet, which means the addition of gas microbubbles enhances the mass transfer process. The quantitative experimental results show the effects of equipment length, flow rates, and phase ratios on the extraction efficiency. kLa is introduced to characterize the mass transfer rate. Within the equipment length of 30 cm, kLa increases with the increase of gas flow rates mostly because of the sharply increased specific area. The kLa value of a gas–liquid–liquid hollow droplet flow increases around 10–60 times compared to that of a liquid–liquid droplet flow system. The length of the extraction equipment needed to reach 95% extraction efficiency reduced around 10–1000 times when gas microbubbles are introduced. Based on the experimental data, a theoretical model was built for the potential prediction of the enhancement of extraction by adding gas microbubbles. An effective diffusion coefficient is introduced to combine the convective mass transfer factor into this model. The modelling results fit well with the experimental data. With this model, some predictions for the enhancement of extraction are made with different flow conditions. All the abovementioned results present a practical method for the enhancement of extraction process with high phase ratio systems, which has potential applications in different areas including analytical chemistry, micro-extraction and biological extraction. For the use of this method, to obtain the stable gas-in-liquid-in-liquid flow condition, the interface tensions of three phases have to meet a particular condition, which was stated by Torza36 in 1969. For those systems that do not meet this condition, if it is allowed, surfactants can be used to adjust the interface tensions. However, for the particular systems that do not meet this condition, nor allow additional surfactant, this method cannot be used because of the thermodynamic limitation.Acknowledgements
The authors gratefully acknowledge the supports of the National Natural Science Foundation of China (21476121, 21322604) and the Tsinghua University Initiative Scientific Research Program (2014z21026) for this study.References
- J. R. Burns and C. Ramshaw, Lab Chip, 2001, 1, 10–15 RSC.
- M. N. Kashid, A. Gupta, A. Renken and L. Kiwi-Minsker, Chem. Eng. J., 2010, 158, 233–240 CrossRef CAS PubMed.
- P. Mary, V. Studer and P. Tabeling, Anal. Chem., 2008, 80, 2680–2687 CrossRef CAS PubMed.
- J. H. Xu, J. Tan, S. W. Li and G. S. Luo, Chem. Eng. J., 2008, 141, 242–249 CrossRef CAS PubMed.
- K. Benz, K. P. Jackel, K. J. Regenauer, J. Schiewe, K. Drese, W. Ehrfeld, V. Hessel and H. Lowe, Chem. Eng. Technol., 2001, 24, 11–17 CrossRef CAS.
- K. K. R. Tetala, J. W. Swarts, B. Chen, A. E. M. Janssen and T. A. van Beek, Lab Chip, 2009, 9, 2085–2092 RSC.
- G. Munchow, S. Hardt, J. P. Kutter and K. S. Drese, Lab Chip, 2007, 7, 98–102 RSC.
- Y. S. Huh, S. J. Jeon, E. Z. Lee, H. S. Park and W. H. Hong, Korean J. Chem. Eng., 2011, 28, 633–642 CrossRef CAS.
- Y. S. Huh, C. M. Jeong, H. N. Chang, S. Y. Lee, W. H. Hong and T. J. Park, Biomicrofluidics, 2010, 4, 014103 CrossRef PubMed.
- M. C. Morales and J. D. Zahn, Microfluid. Nanofluid., 2010, 9, 1041–1049 CrossRef CAS.
- K. H. Nam, W. J. Chang, H. Hong, S. M. Lim, D. I. Kim and Y. M. Koo, Biomed. Microdevices, 2005, 7, 189–195 CrossRef PubMed.
- P. Wagli, Y. C. Chang, A. Homsy, L. Hvozdara, H. P. Herzig and N. F. de Rooij, Anal. Chem., 2013, 85, 7558–7565 CrossRef CAS PubMed.
- P. Znidarsic-Plazl and I. Plazl, Lab Chip, 2007, 7, 883–889 RSC.
- O. Tamagawa and A. Muto, Chem. Eng. J., 2011, 167, 700–704 CrossRef CAS PubMed.
- D. Tsaoulidis and P. Angeli, Chem. Eng. J., 2015, 262, 785–793 CrossRef CAS PubMed.
- C. Priest, J. F. Zhou, S. Klink, R. Sedev and J. Ralston, Chem. Eng. Technol., 2012, 35, 1312–1319 CrossRef CAS PubMed.
- D. Ciceri, L. R. Mason, D. J. E. Harvie, J. M. Perera and G. W. Stevens, Microfluid. Nanofluid., 2013, 14, 213–224 CrossRef CAS.
- T. Maruyama, H. Matsushita, J. Uchida, F. Kubota, N. Kamiya and M. Goto, Anal. Chem., 2004, 76, 4495–4500 CrossRef CAS PubMed.
- H. Shen, Q. Fang and Z. L. Fang, Lab Chip, 2006, 6, 1387–1389 RSC.
- S. A. Bowden, P. B. Monaghan, R. Wilson, J. Parnell and J. M. Cooper, Lab Chip, 2006, 6, 740–743 RSC.
- J. Hereijgers, N. van Oeteren, J. F. M. Denayer, T. Breugelmans and W. De Malsche, Chem. Eng. J., 2015, 273, 138–146 CrossRef CAS PubMed.
- N. D. Raimondi, L. Prat, C. Gourdon and J. Tasselli, Chem. Eng. Sci., 2014, 105, 169–178 CrossRef PubMed.
- C. Priest, M. D. Reid and C. P. Whitby, J. Colloid Interface Sci., 2011, 363, 301–306 CrossRef CAS PubMed.
- Z. Barikbin, M. T. Rahman, P. Parthiban, A. S. Rane, V. Jain, S. Duraiswamy, S. H. S. Lee and S. A. Khan, Lab Chip, 2010, 10, 2458–2463 RSC.
- P. M. Nesterov, V. V. Kafarov and V. V. Shestopa, Ind. Lab., 1967, 33, 83 Search PubMed.
- X. C. Gong, Y. C. Lu, Z. X. Qian and G. S. Luo, Ind. Eng. Chem. Res., 2009, 48, 4507–4513 CrossRef CAS.
- H. Ahmed, H. Diamonta, C. Chaker and R. Abdelhamid, Sep. Purif. Technol., 2007, 55, 212–216 CrossRef PubMed.
- M. I. Amin, M. M. Ali, H. M. Kamal, A. M. Youssef and M. A. Akl, Hydrometallurgy, 2010, 105, 115–119 CrossRef CAS PubMed.
- A. Drelinkiewicz, R. Laitinen, R. Kangas and J. Pursiainen, Appl. Catal., A, 2005, 284, 59–67 CrossRef CAS PubMed.
- L. Xu, C. Basheer and H. K. Lee, J. Chromatogr. A, 2007, 1152, 184–192 CrossRef CAS PubMed.
- J. Tan, Z. D. Liu, Y. C. Lu, J. H. Xu and G. S. Luo, Sep. Purif. Technol., 2011, 80, 225–234 CrossRef CAS PubMed.
- W. T. Wang, R. Chen, J. H. Xu, Y. D. Wang and G. S. Luo, RSC Adv., 2014, 4, 16444–16448 RSC.
- W. T. Wang, R. Chen, J. H. Xu, Y. D. Wang and G. S. Luo, Chem. Eng. J., 2015, 263, 412–418 CrossRef CAS PubMed.
- J. H. Xu, R. Chen, Y. D. Wang and G. S. Luo, Lab Chip, 2012, 12, 2029–2036 RSC.
- R. Chen, P. F. Dong, J. H. Xu, Y. D. Wang and G. S. Luo, Lab Chip, 2012, 12, 3858–3860 RSC.
- S. Torza and S. G. Mason, Science, 1969, 163, 813–814 CrossRef CAS PubMed.
- W. K. Lewis and W. G. Whitman, Ind. Eng. Chem., 1924, 16, 1215–1220 CrossRef CAS.
- M. N. Kashid, A. Renken and L. Kiwi-Minsker, Chem. Eng. Sci., 2011, 66, 3876–3897 CrossRef CAS PubMed.
- H. Brenner, Chem. Eng. Sci., 1963, 18, 109–122 CrossRef CAS.
- L. Oellrich, H. Schmidtt and H. Brauer, Chem. Eng. Sci., 1973, 28, 711–721 CrossRef CAS.
- P. N. Rowe, K. T. Claxton and J. B. Lewis, Trans. Inst. Chem. Eng., 1965, 43, 14–31 Search PubMed.
- C. T. Culbertson, S. C. Jacobson and J. M. Ramsey, Talanta, 2002, 56, 365–373 CrossRef CAS.
- C. R. Wilke and P. Chang, AIChE J., 1955, 1, 264–270 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra15769b |
| This journal is © The Royal Society of Chemistry 2015 |