
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the high reactivity of carbonyl compounds towards nucleophilic carbenoid intermediates generated from carbene isocyanides
Mar Ríos-Gutiérreza,
Luis R. Domingo*a and
Patricia Pérezb
aUniversidad de Valencia, Facultad de Química, Departamento de Química Orgánica, Dr Moliner 50, E-46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es; Web: http://www.luisrdomingo.com
bUniversidad Andrés Bello, Facultad de Ciencias Exactas, Departamento de Ciencias Químicas, Av. República 230, 8370146, Santiago, Chile
First published on 14th September 2015
Abstract
The high reactivity of carbonyl compounds towards the carbenoid intermediate cis-IN, generated in situ by the addition of methyl isocyanide to dimethyl acetylenedicarboxylate (DMAD), has been investigated at the MPWB1K/6-311G(d,p) computational level by using Molecular Electron-Density Theory (MEDT). This multicomponent (MC) reaction is a domino process that comprises two sequential reactions: (i) the formation of a nucleophilic carbenoid intermediate trans-IN; and (ii) the nucleophilic attack of cis-IN on the carbonyl compound, resulting in the formation of the final 2-iminofuran derivative. The present MEDT study establishes that the high nucleophilic character and the electronic structure of the carbenoid intermediate, cis-IN, together with the specific approach mode of the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond during the nucleophilic attack of the sp2 hybridised carbenoid C4 center of cis-IN on the carbonyl C5 carbon of acetone, enables the formation of the C4–C5 single bond with a very low activation enthalpy, 3.3 kcal mol−1, without any external electrophilic activation of the carbonyl group, and the subsequent ring closure through the downhill formation of the C–O single bond. The Bonding Evolution Theory (BET) study for the formation of the 2-iminofuran allows characterisation of the mechanism as a [2n + 2n] cycloaddition, ruling out the proposed 1,3-dipolar cycloaddition mechanism.
O double bond during the nucleophilic attack of the sp2 hybridised carbenoid C4 center of cis-IN on the carbonyl C5 carbon of acetone, enables the formation of the C4–C5 single bond with a very low activation enthalpy, 3.3 kcal mol−1, without any external electrophilic activation of the carbonyl group, and the subsequent ring closure through the downhill formation of the C–O single bond. The Bonding Evolution Theory (BET) study for the formation of the 2-iminofuran allows characterisation of the mechanism as a [2n + 2n] cycloaddition, ruling out the proposed 1,3-dipolar cycloaddition mechanism.
Introduction
Carbene isocyanides I are essential building blocks in modern organic chemistry,1 and it has been reported that these species nucleophilically attack dialkyl acetylenedicarboxylates II yielding zwitterionic species III, which act as crucial intermediates. These reactive intermediates are readily trapped by several kinds of electrophilic carbon molecules2 such as aldehydes,3 ketones,4 esters,5 and sulfonylimines,6 and even carbon dioxide.7 When the electrophilic molecules are carbonyl derivatives IV, the reaction products are 2-iminofuran derivatives V (see Scheme 1). Polyfunctionalised furans are versatile synthetic starting materials for the preparation of a great variety of heterocyclic and acyclic compounds,8 and especially 2,5-disubstituted furan-3,4-dicarboxylates, which are very important starting materials in the synthesis of natural products containing tetrahydrofuran rings.9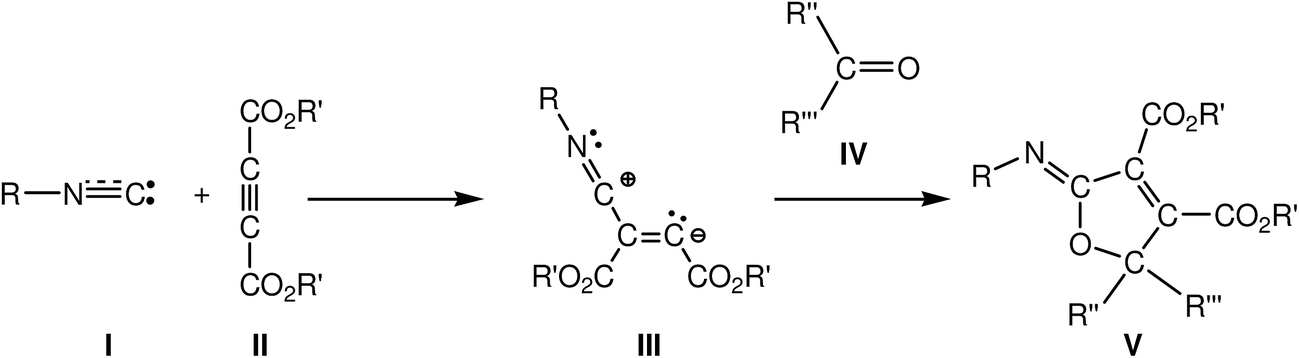 | ||
| Scheme 1 | ||
This multicomponent (MC) reaction is a domino process that comprises two consecutive reactions: (i) a nucleophilic attack of the carbene isocyanides I on the acetylenedicarboxylates II, yielding the proposed zwitterionic intermediates III; and (ii) the quick capture of these intermediates by a carbonyl derivative IV yielding the 2-iminofuran derivatives V. This last step has been associated with a 1,3-dipolar cycloaddition reaction in which the zwitterionic intermediate III participates as the 1,3-dipole and the carbonyl derivative acts as the dipolarophile.3,4
For this 1,3-dipolar cycloaddition reaction two mechanisms have been proposed (see Scheme 2): (i) a one-step mechanism in which the C–C and C–O bonds are formed in a single step but asynchronously;4a and (ii) a stepwise mechanism in which a new zwitterionic intermediate VI is formed through the nucleophilic attack of the zwitterionic intermediate III on the carbonyl derivative IV; the subsequent cyclisation of this intermediate will yield the 2-iminofuran derivative V.4b
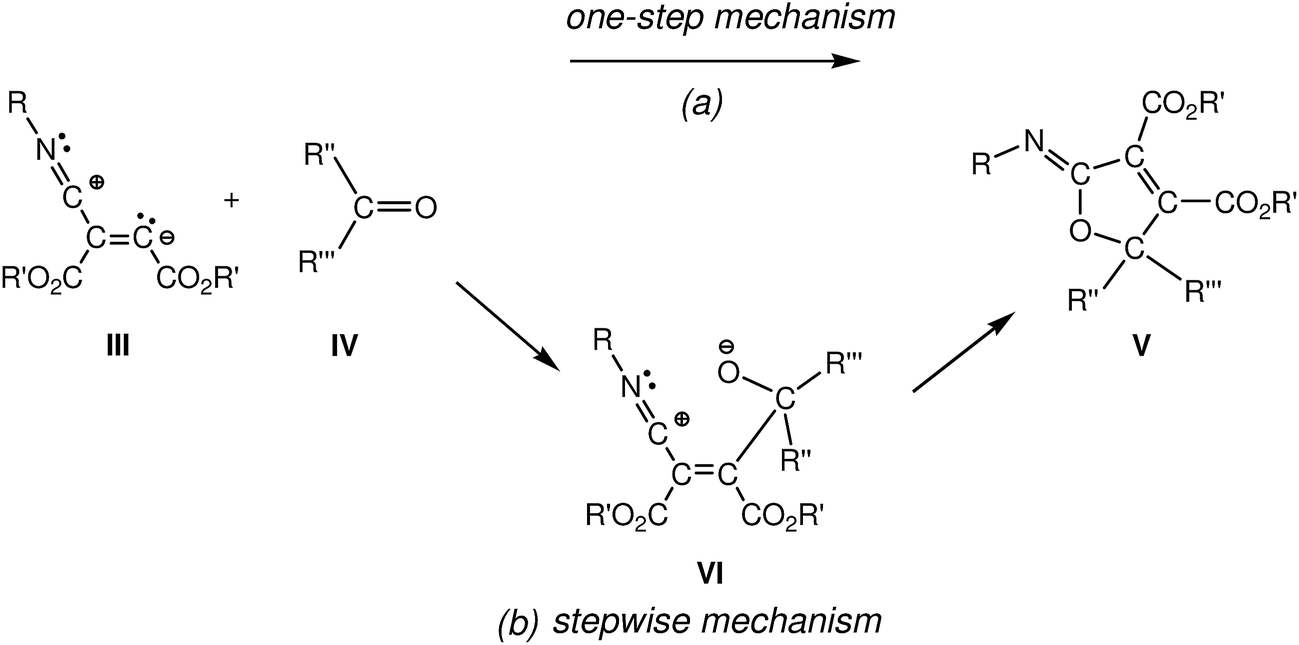 | ||
| Scheme 2 Proposed mechanisms for the 1,3-dipolar cycloadditions of intermediate III with carbonyl derivatives IV. | ||
Due to the high reactivity evidenced by the intermediate III and the significance of the formation of the 2-iminofurans V through these MC reactions involving a large diversity of carbonyl derivatives IV,3–5,7 a Density Functional Theory (DFT) study of the MC reaction between methyl isocyanide 1, DMAD 2 and acetone 3 yielding 2-iminofuran 4 is performed herein, using Molecular Electron-Density Theory (MEDT) to explain this high reactivity (see Scheme 3). The proposed MEDT, in which the changes in the electron-density and not molecular orbital interactions are considered responsible for reactivity in organic chemistry, uses quantum chemical tools based on the analysis of electron-density, such as the conceptual DFT reactivity indices10 and the topological Electron Localisation Function11 (ELF) analysis of the changes in the molecular electron-density along the reaction path, in order to establish the molecular mechanism of an organic reaction.
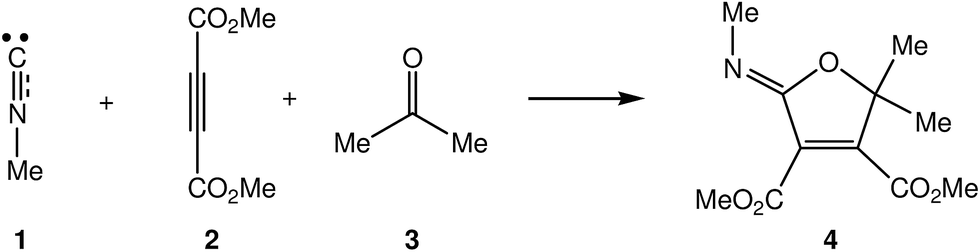 | ||
| Scheme 3 Selected reaction model for the MC reaction of isocyanides I, acetylenedicarboxylate derivatives II and carbonyl compounds IV. | ||
To this end, a theoretical study at the MPWB1K/6-311G(d,p) computational level, in which a combination of (i) the exploration and characterisation of the potential energy surfaces (PESs) associated with the selected domino reaction, (ii) the analysis of the reactivity indices derived from the conceptual DFT for the ground state of the reagents and (iii) the Bonding Evolution Theory12 (BET) analysis of the two consecutive reactions, was carried out. Three unresolved questions will be answered: (i) what is the electronic structure of the intermediate III formed in these domino reactions?; (ii) what is the origin of the high reactivity of the carbonyl compounds IV in these MC reactions?; and finally, (iii) what is the mechanism of the cycloaddition step? As these questions cannot be experimentally resolved, our theoretical study provides valuable information about these MC reactions.
Computational methods
Several studies have shown that the B3LYP functional13 is relatively accurate for providing kinetic data, although the reaction exothermicities are underestimated.14 Truhlar’s group has proposed some functionals, such as the MPWB1K functional,15 which gives good results for combinations of thermochemistry, thermochemical kinetics and other weak interactions. Therefore, in this study the MPWB1K functional was selected together with the standard 6-31G(d) basis set.16 The optimisations were carried out using the Berny analytical gradient optimisation method.17 The stationary points were characterised by frequency computations in order to verify that the TSs have one and only one imaginary frequency. The IRC paths18 were traced in order to check the energy profiles connecting each TS to the two associated minima in the proposed mechanism using the second order González–Schlegel integration method.19 Solvent effects were taken into account by full optimisation of the gas phase structures at the MPWB1K/6-311G(d,p) level using the polarisable continuum model (PCM) developed by Tomasi’s group20 in the framework of the self-consistent reaction field (SCRF).21 The integral equation formalism variant is the SCRF method used in this work. Several polar solvents such as dichloromethane, benzene and acetonitrile have been used in these MC reactions. Acetonitrile was selected for the solvent effect calculations since it was used in the reactions of a benzo[b]acridine-6,11-dione acting as the carbonyl derivative.4b The values of enthalpy, entropy and Gibbs free energy in acetonitrile were calculated with the standard statistical thermodynamics at 25 °C and 1 atm.16 No scaling factor in the thermodynamic calculations was used. The electronic structures of stationary points were characterised by natural bond orbital (NBO) analysis.22Conceptual DFT23 provides different indices to rationalize and understand chemical structure and reactivity.10 In this sense, the global electrophilicity index,24 ω, is given by the following expression, ω = (μ2/2η), in terms of the electronic chemical potential μ and the chemical hardness η. Both quantities may be approached in terms of the one-electron energies of the frontier molecular orbitals HOMO and LUMO, εH and εL, as μ ≈ (εH + εL)/2 and η ≈ (εL − εH), respectively.25 The empirical (relative) nucleophilicity index,26 N, based on the HOMO energies obtained within the Kohn–Sham scheme,27 is defined as N = EHOMO(Nu) − EHOMO(TCE), where tetracyanoethylene (TCE) is the reference because it presents the lowest HOMO energy in a long series of molecules already investigated in the context of polar organic reactions. This choice allows convenient handling of a nucleophilicity scale of positive values. Nucleophilic Pk− and electrophilic Pk+ Parr functions28 were obtained through the analysis of the Mulliken atomic spin density of the corresponding radical cations or anions.
The characterisation of the electron-density reorganisation to evidence the bonding changes along a reaction path is the most attractive method to characterise a reaction mechanism.29 To perform these analyses quantitatively, the BET,12 consisting of the joint-use of ELF topology and Thom’s catastrophe theory30 (CT), proposed by Krokidis et al.,12 is a new tool for analysing the electronic changes in chemical processes. BET has been applied to different elementary reactions,31 allowing the molecular mechanism to be established. The ELF topological analysis, η(r),32 was performed with the TopMod program33 using the corresponding MPWB1K/6-311G(d,p) monodeterminantal wavefunctions of the selected structures of the IRC. Non-covalent interactions (NCIs) were computed using the methodology previously described.34 All computations were carried out with the Gaussian 09 suite of programs.35
Results and discussion
The present theoretical study has been divided into four parts: (i) first, the PESs associated with the domino reaction between isocyanide 1, DMAD 2 and acetone 3 yielding 2-iminofuran 4 are explored and characterised; (ii) then, an analysis of the DFT reactivity indices for the ground state of the reagents involved in this domino process is carried out; (iii) next, a BET study of the two consecutive reactions is performed in order to characterise the molecular mechanism; and finally, (iv) the electronic structure of the intermediate cis-IN as well as the origin of the high reactivity of carbonyl compounds in these MC reactions are discussed.(i) Study of the PESs of the MC reaction between methyl isocyanide 1, DMAD 2 and acetone 3
The MC reaction between methyl isocyanide 1, DMAD 2 and acetone 3 to yield 2-iminofuran 4 is a domino reaction that comprises two addition reactions (see Scheme 4). The first one is the nucleophilic attack of the carbene methyl isocyanide 1 on one of the two electrophilic acetylenic carbon atoms of DMAD 2 to yield the intermediate trans-IN, which after isomerisation to the intermediate cis-IN attacks acetone 3. The total and relative electronic energies of the stationary points involved in the two consecutive processes of the MC reaction between isocyanide 1, DMAD 2 and acetone 3 are given in Table 1.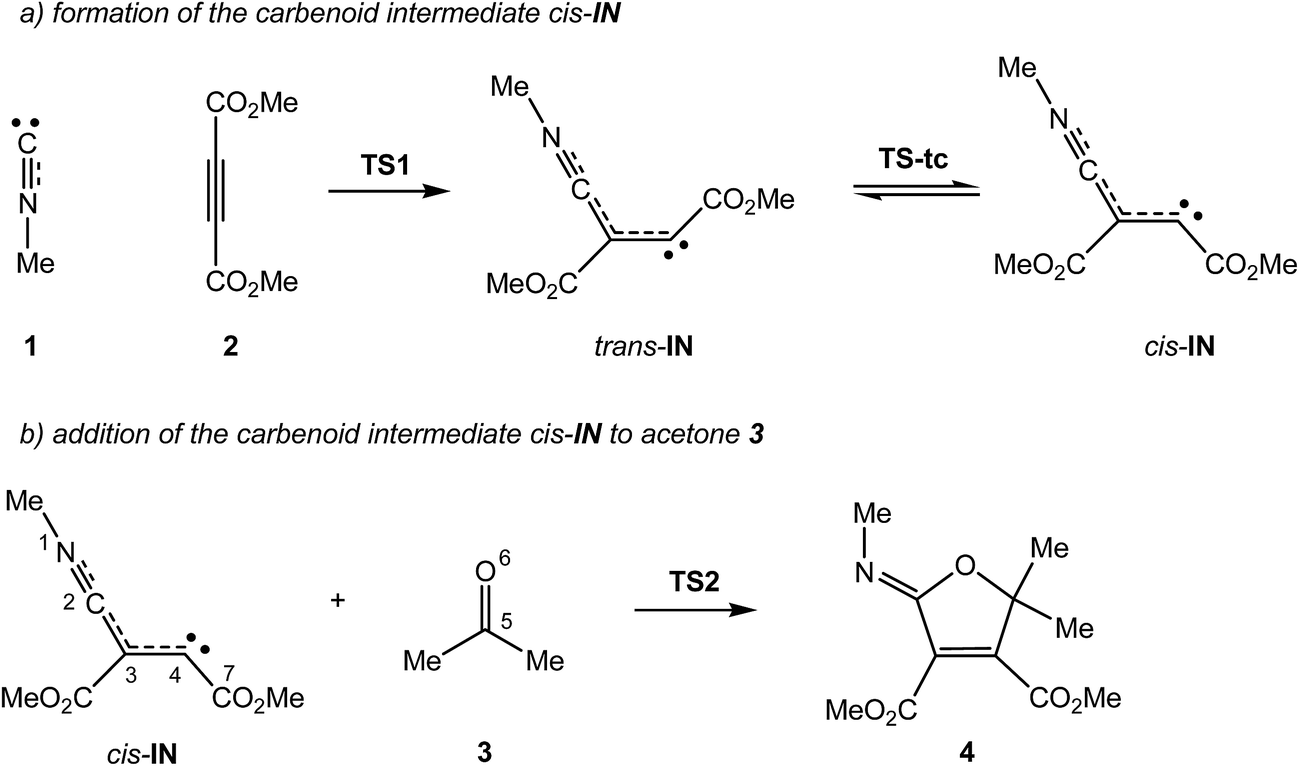 | ||
| Scheme 4 Domino reaction between methyl isocyanide 1, DMAD 2 and acetone 3. | ||
| Gas phase | Acetonitrile | |||
|---|---|---|---|---|
| E | ΔE | E | ΔE | |
| a Relative to 1, 2 and 3. | ||||
| 1 | −132.674951 | −132.681758 | ||
| 2 | −532.962501 | −532.971986 | ||
| 3 | −193.098437 | −193.104691 | ||
| TS1 | −665.609778 | 17.4 | −665.624629 | 18.3 |
| trans-IN | −665.638068 | −0.4 | −665.664217 | −6.6 |
| TS-tc | −665.622520 | 9.4 | −665.649492 | 2.7 |
| cis-IN | −665.639621 | −1.4 | −665.666500 | −6.6 |
| TS2 | −858.744588 | −5.5 | −858.765118 | −4.2 |
| 4 | −858.876561 | −88.3 | −858.888753 | −81.8 |
The activation energy associated with the nucleophilic attack of carbene methyl isocyanide 1 on DMAD 2, via TS1, presents a high value, 17.4 kcal mol−1, with the formation of the intermediate trans-IN being slightly exothermic, −0.4 kcal mol−1. This intermediate, which presents a trans disposition of the two carboxylate groups, undergoes a sp2–sp–sp2 rehybridisation process at the C4 carbon via TS-tc with an estimated activation energy of 9.4 kcal mol−1 to yield cis-IN, which is 1.4 kcal mol−1 lower in energy than trans-IN. It is noteworthy that the cis rearrangement of the two carboxylate groups at cis-IN is required for the subsequent addition of acetone 3 followed by the concomitant ring closure. From this intermediate, the formation of 2-iminofuran 4 takes place through the nucleophilic attack of cis-IN on acetone 3, followed by a downhill C–O bond formation. This nucleophilic attack via TS2 presents a very low activation energy, 4.1 kcal mol−1, the cycloaddition being strongly exothermic by 88.3 kcal mol−1.
The activation and reaction energies associated with TS1 and TS2, and 2-iminofuran 4, decrease by 1 kcal mol−1 and 6 kcal mol−1, respectively, when the solvent effects of acetonitrile are considered. On the contrary, the activation and reaction energies associated with TS-tc, and trans-IN and cis-IN, increase by between 5–7 kcal mol−1. This behaviour is a consequence of the higher solvation of the intermediates trans-IN and cis-IN than of the reagents, TSs, and the final product for this MC reaction.
The relative and non-relative enthalpies, entropies and Gibbs free energies of the stationary points involved in the domino reaction between methyl isocyanide 1, DMAD 2 and acetone 3 are displayed in Table 2. The Gibbs free energy profile including both consecutive processes is graphically represented in Fig. 1. Inclusion of the thermal corrections in the electronic energies does not significantly change the relative enthalpies of the stationary points involved in this MC reaction; those of TS1, TS-tc and cis-IN remain almost unchanged but those of trans-IN, TS2 and 4 slightly increase by 1–5 kcal mol−1. Inclusion of the entropies in the enthalpies increases the relative Gibbs free energies by between 11 and 33 kcal mol−1, due to the unfavourable entropy associated with this bimolecular reaction.
| H | ΔH | S | ΔS | G | ΔG | |
|---|---|---|---|---|---|---|
| a Relative to 1, 2 and 3. | ||||||
| 1 | −132.630638 | 58.298 | −132.658324 | |||
| 2 | −532.841793 | 104.873 | −532.891597 | |||
| 3 | −193.013050 | 71.406 | −193.046960 | |||
| TS1 | −665.442939 | 18.5 | 127.102 | −36.1 | −665.503299 | 29.3 |
| trans-IN | −665.481317 | −5.6 | 120.401 | −42.8 | −665.538495 | 7.2 |
| TS-tc | −665.467932 | 2.8 | 118.198 | −45.0 | −665.524063 | 16.2 |
| cis-IN | −665.482621 | −6.4 | 125.618 | −37.6 | −665.542277 | 4.8 |
| TS2 | −858.488597 | −2.0 | 150.779 | −83.8 | −858.560201 | 23.0 |
| 4 | −858.607120 | −76.3 | 143.575 | −91.0 | −858.675303 | −49.2 |
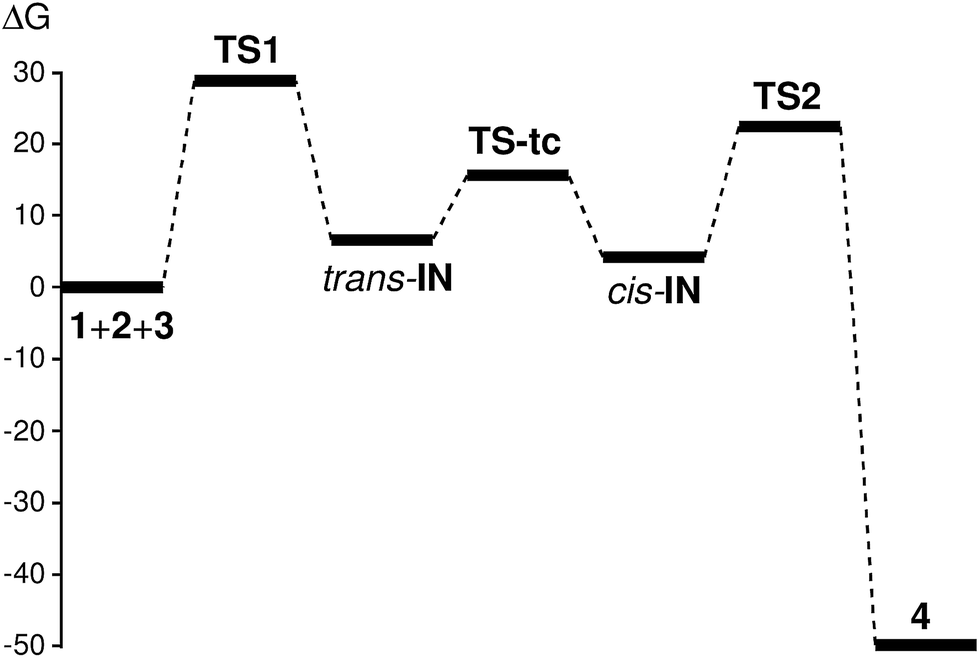 | ||
| Fig. 1 Gibbs free energy profile (ΔG, in kcal mol−1) of the domino reaction between methyl isocyanide 1, DMAD 2 and acetone 3. | ||
Thus, the activation Gibbs free energy associated with the first nucleophilic attack of isocyanide 1 on DMAD 2, via TS1, is 29.3 kcal mol−1, the formation of the intermediate trans-IN being endergonic by 7.2 kcal mol−1. The following conversion of trans-IN into cis-IN presents an activation Gibbs free energy of 9.0 kcal mol−1 (TS-tc), the conversion being exergonic by 2.4 kcal mol−1. Finally, the cycloaddition reaction of cis-IN with acetone 3 via TS2 yields 2-iminofuran 4 with an activation Gibbs free energy of 23.0 kcal mol−1. The formation of the final cycloadduct 4 is strongly exergonic by 49.2 kcal mol−1.
Some interesting conclusions can be drawn from the Gibbs free energy profile presented in Fig. 1: (i) the first nucleophilic attack of carbene isocyanide 1 on DMAD 2 is the rate-determining step (RDS) of this domino process; (ii) the intermediate trans-IN quickly isomerises to cis-IN, which is 2.4 kcal mol−1 more stable and whose cis disposition is required for the following cycloaddition reaction with acetone 3 to occur; (iii) the activation Gibbs free energy of the cycloaddition reaction of the intermediate cis-IN with acetone 3 via TS2 is 6.3 kcal mol−1 lower than that of the RDS of the domino process via TS1; (iv) the strong exergonic character of the cycloaddition reaction between cis-IN and acetone 3 makes this process irreversible; and (v) although the formation of the intermediate cis-IN is slightly endergonic, as soon as it is formed it is quickly and irreversibly captured by acetone 3 yielding 2-iminofuran 4. Accordingly, the MC reaction between isocyanide 1, DMAD 2 and acetone 3 is kinetically and thermodynamically very favourable.
The geometries of the TSs involved in the domino reaction between methyl isocyanide 1, DMAD 2 and acetone 3 in acetonitrile are shown in Fig. 2. At TS1, associated with the nucleophilic addition of methyl isocyanide 1 to DMAD 2, the length of the C2–C3 forming bond is 1.973 Å, while at the intermediates trans-IN and cis-IN the length of the formed C2–C3 single bond is 1.407 Å. At TS2, associated with the cycloaddition reaction of cis-IN with acetone 3, the lengths of the C4–C5 and C2–O6 forming bonds are 2.147 and 2.556 Å. The IRC from TS2 towards the final 2-iminofuran 4 indicates that the second reaction of this MC process is associated with a two-stage one-step mechanism36 in which the C4–C5 single bond is completely formed before the formation of the second C2–O6 single bond starts (see later). In addition, this IRC also shows that the carbonyl C5–O6 bond of acetone 3 approaches the intermediate cis-IN in the C2–C3–C4 plain, in which the two C4–C5 and C2–O6 single bonds will be formed (see TS2 in Fig. 2). This approach mode is different to that demanded in 1,3-dipolar cycloadditions, in which the dipolarophile approaches over the plain of the 1,3-dipole.
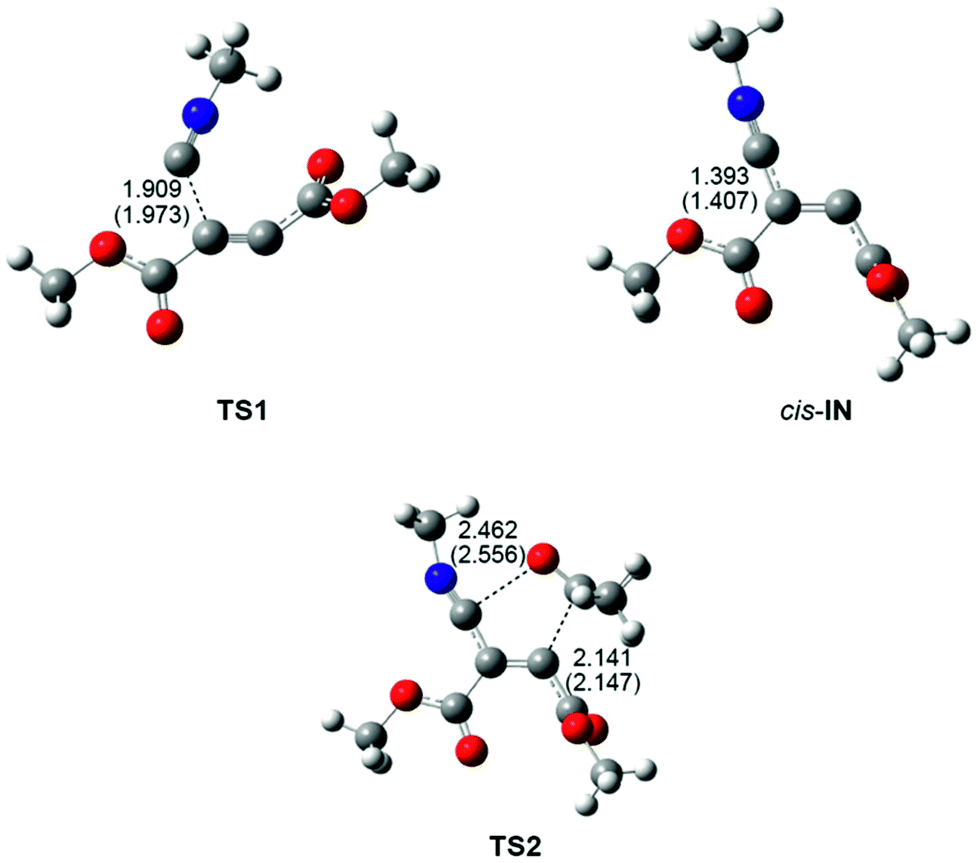 | ||
| Fig. 2 MPWB1K/6-311G(d,p) geometries of the most important stationary points involved in the domino reaction between methyl isocyanide 1, DMAD 2 and acetone 3. Distances are given in Angstroms. Lengths in acetonitrile are given in parentheses. | ||
The electronic nature of the nucleophilic addition reaction of carbene isocyanide 1 with DMAD 2, as well as the cycloaddition reaction of the intermediate cis-IN with acetone 3, were analysed by computing the global electron-density transfer (GEDT).37 The natural atomic charges, obtained through a natural population analysis (NPA), were shared between the two frameworks involved in these addition reactions. Thus, the GEDT that fluxes from the carbene isocyanide framework towards the acetylene derivative during the first reaction is 0.26e at TS1 and 0.62e at trans-IN, indicating that during the nucleophilic addition of isocyanide 1 to DMAD 2 there is an increase of the GEDT until the maximum value is reached with the formation of the C2–C3 single bond at the intermediate trans-IN. On the other hand, the GEDT that fluxes from the intermediate cis-IN to the ketone framework during the second process is 0.26e at TS2, showing the polar character of the cycloaddition reaction between cis-IN and acetone 3. The GEDT values calculated for both reactions emphasise the polar nature of the MC reaction between methyl isocyanide 1, DMAD 2 and acetone 3.
(ii) Analysis of the global and local DFT reactivity indices for the ground state of the reagents and of intermediate cis-IN
Studies devoted to polar organic reactions have shown that the analysis of the reactivity indices defined within conceptual DFT10 is a powerful tool to understand the reactivity in polar cycloadditions. The global DFT indices, namely, the electronic chemical potential μ, chemical hardness η, global electrophilicity ω and nucleophilicity N, of methyl isocyanide 1, DMAD 2, acetone 3 and the intermediate cis-IN are given in Table 3.| μ | η | ω | N | |
|---|---|---|---|---|
| DMAD 2 | −5.01 | 8.95 | 1.40 | 0.91 |
| cis-IN | −3.58 | 6.03 | 1.06 | 3.80 |
| Acetone 3 | −3.72 | 9.02 | 0.77 | 2.16 |
| Methyl isocyanide 1 | −3.90 | 11.46 | 0.66 | 0.77 |
| Acetylene 5 | −3.53 | 11.34 | 0.55 | 1.20 |
The electronic chemical potential of carbene isocyanide 1, μ = −3.90 eV, is higher than that of DMAD 2, μ = −5.01 eV, indicating that for a polar reaction the GEDT33 will flux from the carbene isocyanide framework towards the electron-deficient acetylene framework. In the same way, the higher electronic chemical potential of the intermediate cis-IN, μ = −3.58 eV, than that of acetone 3, μ = −3.72 eV, suggests that for the subsequent cycloaddition reaction between the intermediate cis-IN and acetone 3, the GEDT will flux towards the ketone framework.
Methyl isocyanide 1 presents an electrophilicity ω index of 0.66 eV and a nucleophilicity N index of 0.77 eV, being classified on the borderline of moderate electrophiles38 and as a marginal nucleophile.39 Accordingly, carbene isocyanide 1 is considered a weak nucleophile participating in polar reactions and, therefore, a strongly electrophilically activated molecule will be necessary to make the nucleophilic attack of methyl isocyanide 1 feasible.
Polar organic reactions require the participation of good electrophiles and good nucleophiles. Acetylene 5 is one of the poorest electrophilic, ω = 0.55 eV, and nucleophilic, N = 1.20 eV, species involved in polar organic reactions, being classified as a marginal electrophile and a marginal nucleophile. Therefore, it cannot participate in polar reactions. The inclusion of two methyl carboxylate groups in the acetylene framework drastically increases the electrophilicity ω index of DMAD 2, ω = 1.40 eV, being classified as a strong electrophile, and slightly decreases its nucleophilicity N index to N = 0.91 eV, remaining classified as a marginal nucleophile. In spite of the strong electrophilic character of DMAD 2, the low nucleophilic character of isocyanide 1 accounts for the high activation energy associated with the nucleophilic addition of isocyanide 1 to DMAD 2 (see above).
Otherwise, the electrophilicity ω and nucleophilicity N indices of the intermediate cis-IN, ω = 1.06 eV and N = 3.80 eV, allow its classification on the borderline of strong electrophiles and as a strong nucleophile. Acetone 3, which presents an electrophilicity ω index of ω = 0.77 eV and a nucleophilicity N index of N = 2.16 eV, will behave as a moderate electrophile and a moderate nucleophile. It is interesting to note that the low electrophilic character of acetone 3 demands its electrophilic activation in order for it to participate in polar reactions.
Recently, the electrophilic Pk+ and nucleophilic Pk− Parr functions have been proposed to analyse the local reactivity in polar processes28 involving reactions between a nucleophile–electrophile pair. Accordingly, the electrophilic Pk+ Parr functions for DMAD 2 and acetone 3, and the nucleophilic Pk− Parr functions for methyl isocyanide 1 and the intermediate cis-IN are analysed (see Fig. 3).
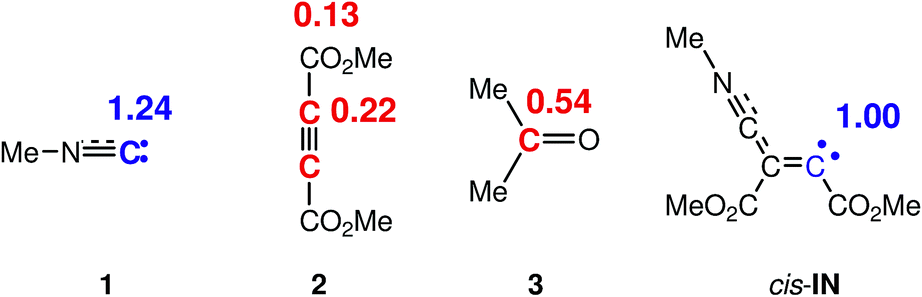 | ||
| Fig. 3 Nucleophilic Pk− Parr functions, in blue, and electrophilic Pk+ Parr functions, in red, in methyl isocyanide 1, DMAD 2, acetone 3 and intermediate cis-IN. | ||
The analysis of the nucleophilic Pk− Parr functions of carbene isocyanide 1 and cis-IN shows that the C2 carbon of isocyanide 1 and the C4 carbon of the intermediate cis-IN present the maximum values, Pk− = 1.24 and 1.00, respectively, indicating that these sites are the most nucleophilic centers of these species (see Scheme 4 for atom numbering). From these values two appealing conclusions can be obtained: (i) carbene isocyanide 1 experiences a strong nucleophilic activation at the C2 carbon; and (ii) for the nucleophilic intermediate cis-IN, the nucleophilic Pk− Parr functions are concentrated at the C4 carbon. The strong nucleophilic activation at the C2 carbon of isocyanide 1 is a consequence of the nucleophilic deactivation of the N1 nitrogen atom.
On the other hand, analysis of the electrophilic Pk+ Parr functions of DMAD 2 indicates that the acetylene C3 and C4 carbons, Pk+ = 0.22, are ca. twice as electrophilically activated as the carbonyl carbons, Pk+ = 0.13. Finally, acetone 3 presents its electrophilic activation at the carbonyl carbon atom, Pk+ = 0.54.
Consequently, the most favourable electrophile–nucleophile interaction for the nucleophilic attack of carbene isocyanide 1 on DMAD 2 will take place between the most nucleophilic center of isocyanide 1, the C2 carbon, and the most electrophilic center of DMAD 2, the C3 or C4 carbon. Likewise, the most favourable bond formation for the nucleophilic attack of the intermediate cis-IN on acetone 3 will take place between the most nucleophilic center of the former, the C4 carbon, and the electrophilic carbon atom of acetone 3.
(iii) BET analysis of the domino reaction between methyl isocyanide 1, DMAD 2 and acetone 3
Several theoretical studies have shown that the ELF topological analysis of the changes of electron-density, ρ(r), along a reaction path can be used as a valuable tool to understand the bonding changes along the reaction path, and consequently to establish the molecular mechanisms.29 After an analysis of the electron-density, ELF analysis provides basins of attractors, which are the domains in which the probability of finding an electron pair is maximal.40 The spatial points in which the gradient of the ELF has a maximum value are designated as attractors. The basins are classified as core basins and valence basins. The latter are characterised by the synaptic order, i.e. the number of atomic valence shells in which they participate. Thus, there are monosynaptic, disynaptic, trisynaptic basins and so on.41 Monosynaptic basins, labelled V(A), correspond to the lone pairs or non-bonding regions, while disynaptic basins, labelled V(A,B), connect the core of two nuclei A and B and, thus, correspond to a bonding region between A and B. This description recovers the Lewis bonding model, providing a very suggestive graphical representation of the molecular system.ELF topological analysis of the structures involved in an elementary step mainly allows characterisation of three types of valence basins: (i) protonated basins, V(A,H); (ii) monosynaptic basins, V(A), associated with lone pairs or non-bonding regions; and (iii) disynaptic basins, V(A,B), associated with bonding regions. A set of ELF valence basins topologically characterises a molecular structure. Analysis of changes in the number or type of valence basins of the structures involved along the IRC of the reaction allows the establishment of a set of points, Pi, defining the different phases that topologically characterise a molecular mechanism. The further analysis of the different phases characterised by these significant points permits its characterisation.31
Herein, a BET study of the MC reaction between isocyanide 1, DMAD 2 and acetone 3 is performed in order to gain insight into how the bonding changes take place during this domino reaction, and thus, to establish the molecular mechanism of the two consecutive reactions.
| Phases | I | II | III | IV | V | VI | trans-IN | cis-IN |
|---|---|---|---|---|---|---|---|---|
| MC1 | P1 | P2 | P3 | P4 | P5 | |||
| d(C2–C3) | 3.726 | 2.282 | 2.033 | 1.897 | 1.551 | 1.399 | 1.396 | 1.393 |
| GEDT | 0.00 | 0.07 | 0.18 | 0.27 | 0.54 | 0.60 | 0.62 | 0.65 |
| V(N1,C2) | 3.81 | 3.15 | 5.12 | 5.13 | 4.71 | 4.40 | 5.52 | 3.19 |
| V′(N1,C2) | 1.29 | 2.00 | 2.32 | |||||
| V(C3,C4) | 2.68 | 2.37 | 2.23 | 2.10 | 1.73 | 3.10 | 3.13 | 3.14 |
| V′(C3,C4) | 2.65 | 2.48 | 2.16 | 2.02 | 1.74 | |||
| V(C2) | 2.65 | 2.58 | 2.52 | |||||
| V(C2,C3) | 2.55 | 2.66 | 2.73 | 2.71 | 2.65 | |||
| V(C4) | 0.52 | 1.06 | 1.31 | 1.75 | 1.90 | 1.97 | 2.06 | |
| V(N1) | 0.66 | 1.20 |
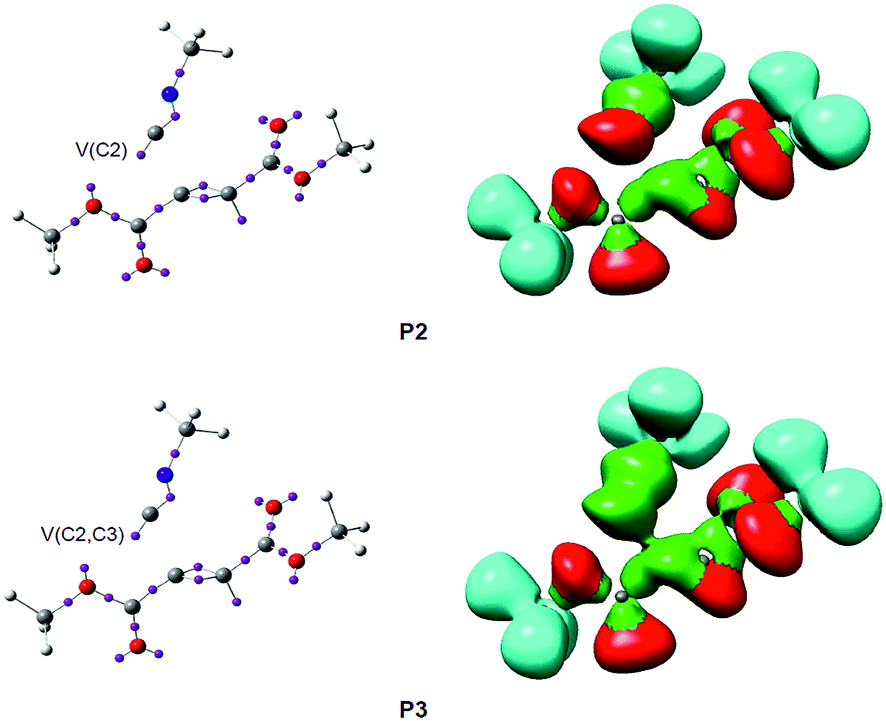 | ||
| Fig. 4 ELF attractor positions and basins for the most relevant points along the IRC associated with the formation of the C2–C3 single bond during the nucleophilic addition reaction of methyl isocyanide 1 with DMAD 2. Disynaptic basins are coloured in green and monosynaptic basins are in red. | ||
 | ||
| Fig. 5 Graphical representation of the basin population changes during the cycloaddition reaction between methyl isocyanide 1 and DMAD 2. Point dotted curves in grey represent the sum of disynaptic basins describing a bond region or monosynaptic basins describing lone pairs. | ||
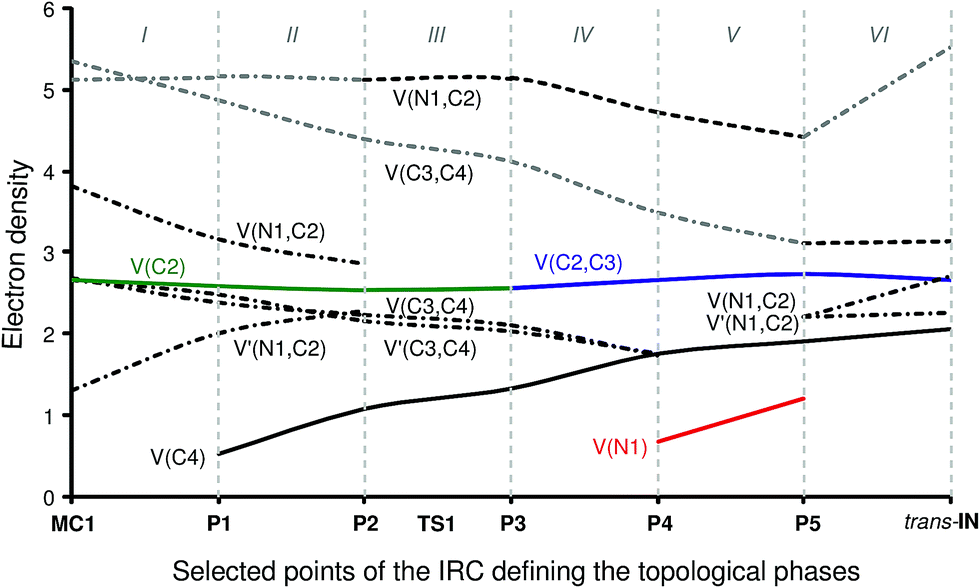
Phase I, 3.73 Å ≥ d(C2–C3) > 2.28 Å, begins at the molecular complex MC1, d(C2–C3) = 3.726 Å, being a minimum in the PES connecting TS1 with the separated reagents 1 and 2. The ELF picture of MC1 exhibits the topological characteristics of the separated reagents. ELF analysis of MC1 shows two V(N1,C2) and V′(N1,C2) disynaptic basins with a population of 3.81e and 1.29e, associated with the N1![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C2 triple bond region of the isocyanide framework, and one V(C2) monosynaptic basin integrating for 2.65e related to the C2 carbon lone pair. In addition, the ELF topology of MC1 also shows the presence of two V(C3,C4) and V′(C3,C4) disynaptic basins with populations of 2.41e and 2.46e belonging to the C3C4 triple bond of the acetylene framework.
C2 triple bond region of the isocyanide framework, and one V(C2) monosynaptic basin integrating for 2.65e related to the C2 carbon lone pair. In addition, the ELF topology of MC1 also shows the presence of two V(C3,C4) and V′(C3,C4) disynaptic basins with populations of 2.41e and 2.46e belonging to the C3C4 triple bond of the acetylene framework.
Phase II, 2.28 Å ≥ d(C2–C3) > 2.03 Å, starts at P1. The first noticeable topological change along the IRC occurs in this phase; a new V(C4) monosynaptic basin, integrating for 0.53e, is created at P1. The electron-density of this basin mainly proceeds from the depopulation of the C3C4 triple bond region until the V(C3,C4) and V′(C3,C4) disynaptic basins reach a total population of 4.85e. Note that the new V(C4) monosynaptic basin is associated with the non-bonding sp2 hybridised lone pair present at the C4 carbon of the nucleophilic intermediate cis-IN. On the other hand, the population of the V(C2) monosynaptic basin slightly decreases. The GEDT has increased to a small extent, 0.07e.
Phase III, 2.03 Å ≥ d(C2–C3) > 1.90 Å, begins at P2. At this point, the two V(N1,C2) and V′(N1,C2) disynaptic basins present at MC1 have merged into a new V(N1,C2) disynaptic basin integrating for 5.12e. This topological change is simply the consequence of electron-density redistribution in the N1–C2 bonding region. In this phase, the TS of the reaction, TS1, d(C2–C3) = 1.909 Å, is found. For this structure, only scanty changes in the electron-density distribution with respect to those found at P1 are observed with the exception that the population of the V(C4) monosynaptic basin has increased to 1.06e. The GEDT has increased to 0.18e.
At phase IV, 1.90 Å ≥ d(C2–C3) > 1.55 Å, which begins at P3, the most significant topological change along the reaction path takes place. The V(C2) monosynaptic basin present at P2 is converted into a new V(C2,C3) disynaptic basin with an initial population of 2.55e (see P2 and P3 in Fig. 4 and the change from V(C1), in green in P2, to V(C2,C3), in blue in P3, in Fig. 5). In spite of the unexpected position of the V(C1) ELF attractor, the depiction of the valence basins associated with P3 shows the disynaptic character of the corresponding basin (see P3 in Fig. 4). This relevant topological change indicates that the formation of the new C2–C3 single bond has already begun at a distance of 1.90 Å with a high electron-density population. In addition, the V(C4) monosynaptic basin increases its population to 1.31e together with the decrease of the total population of the V(C3,C4) and V′(C3,C4) disynaptic basins to 4.12e, which indicates that the C3–C4 bonding region has just acquired its double bond character. At P3, the GEDT is 0.27e.
Phase V, 1.55 Å ≥ d(C2–C3) > 1.40 Å, starts at P4. At this point a new V(N1) monosynaptic basin appears with a population of 0.66e as a consequence of the depopulation of the V(N1,C2) disynaptic basin to 4.71e. The electron-density of the V(C3,C4) and V′(C3,C4) disynaptic basins continues to decrease until it reaches a population of 3.47e, while the V(C4) monosynaptic basin has increased to 1.75e. The GEDT significantly increases to 0.54e.
Finally, the extremely short phase VI, 1.40 Å ≥ d(C2–C3) ≥ 1.39 Å, begins at P5 and ends at the nucleophilic intermediate trans-IN, d(C2–C3) = 1.396 Å. At P5, the two V(C3,C4) and V′(C3,C4) disynaptic basins merge into one V(C3,C4) disynaptic basin integrating for 3.10e, whereas the V(C4) monosynaptic basin has received almost the population of a lone pair, 1.90e. Besides, while the V(N1,C2) disynaptic basin decreases by 0.31e, the V(N1) monosynaptic and V(C2,C3) disynaptic basins increase their populations to 1.20e and 2.73e, respectively. At P5, the maximum GEDT for the reaction takes place, 0.60e. From P5 to trans-IN, the most noticeable topological change is the disappearance of the V(N1) monosynaptic basin simultaneously to the significant increase of the population of the V(N1,C2) disynaptic basin to 5.52e. At trans-IN, the V(C3,C4) disynaptic basin presents a population of 3.10e, indicating that the C3–C4 bonding region is very polarised towards the C2–C3 single bond created at phase IV, which is characterised by one V(C2,C3) disynaptic basin integrating for 2.71e. Finally, the V(C4) monosynaptic basin associated with the C4 carbon lone pair has a population of 1.97e. The GEDT computed at trans-IN, 0.62e, is very high.
| Phases | I | II | III | IV | V | VI | VII | VIII | 4 |
|---|---|---|---|---|---|---|---|---|---|
| MC2 | P1 | P2 | P3 | P4 | P5 | P6 | P7 | ||
| d(C4–C5) | 3.687 | 2.798 | 2.338 | 2.141 | 2.070 | 1.908 | 1.645 | 1.578 | 1.502 |
| d(C2–O6) | 3.090 | 3.007 | 2.623 | 2.462 | 2.412 | 2.288 | 1.953 | 1.746 | 1.370 |
| GEDT | 0.00 | 0.04 | 0.16 | 0.26 | 0.31 | 0.40 | 0.39 | 0.32 | 0.19 |
| V(N1,C2) | 5.50 | 1.78 | 5.48 | 5.53 | 4.75 | 2.14 | 1.83 | 1.72 | 1.64 |
| V′(N1,C2) | 3.70 | 2.09 | 1.84 | 1.74 | 1.63 | ||||
| V(N1) | 0.8 | 1.48 | 2.26 | 2.48 | 2.67 | ||||
| V(C2,C3) | 2.60 | 2.63 | 2.6 | 2.59 | 2.61 | 2.6 | 2.47 | 2.39 | 2.34 |
| V(C3,C4) | 3.17 | 3.16 | 3.24 | 3.27 | 1.68 | 1.64 | 1.71 | 1.73 | 1.73 |
| V′(C3,C4) | 1.61 | 1.68 | 1.68 | 1.68 | 1.69 | ||||
| V(O6) | 2.64 | 2.65 | 2.71 | 2.85 | 2.82 | 2.94 | 2.92 | 2.71 | 2.24 |
| V′(O6) | 2.64 | 2.70 | 2.77 | 2.81 | 2.87 | 2.96 | 2.99 | 2.69 | 2.52 |
| V(C5,O6) | 2.39 | 2.41 | 2.23 | 2.18 | 2.06 | 1.77 | 1.53 | 1.41 | 1.43 |
| V(C4) | 2.05 | 2.00 | 1.92 | ||||||
| V(C4,C5) | 1.87 | 1.89 | 1.94 | 2.05 | 2.06 | 2.09 | |||
| V(C2) | 0.20 | ||||||||
| V(C2,O6) | 0.75 | 1.49 |
 | ||
| Fig. 6 ELF attractor positions for the most relevant points along the IRC associated with the formation of the C4–C5 and C2–O6 single bonds during the cycloaddition reaction of intermediate cis-IN with acetone 3. | ||
 | ||
| Fig. 7 Graphical representation of the basin population changes during the cycloaddition reaction between nucleophilic intermediate cis-IN and acetone 3. Point dotted curves in grey represent the sum of disynaptic basins describing a bond region or monosynaptic basins describing lone pairs. | ||
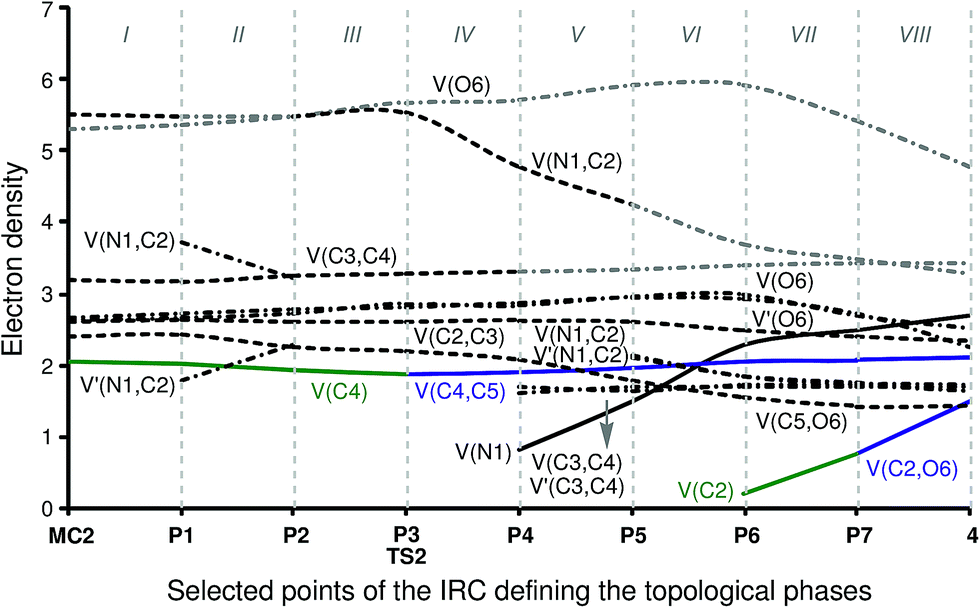
Phase I, 3.69 Å ≥ d(C4–C5) > 2.80 Å and 3.09 Å ≥ d(C2–O6) > 3.00 Å, begins at the molecular complex MC2, d(C4–C5) = 3.687 Å and d(C2–O6) = 3.090 Å, which is a minimum in the PES connecting the intermediate cis-IN and acetone 3 with the corresponding TS2. The ELF picture of MC2 shows the topological behaviour of the separated reagents. Three disynaptic basins, V(N1,C2), V(C2,C3) and V(C3,C4) integrating for 5.50e, 2.60e and 3.17e, respectively, can be observed, which are associated with the N1–C2–C3–C4 bonding region of the intermediate cis-IN. The most important characteristic of this framework is the presence of one V(C4) monosynaptic basin with a population of 2.05e, which is associated with the non-bonding sp2 hybridised lone pair present on the C4 carbon. On the other hand, the ELF analysis of MC2 also shows the presence of two monosynaptic basins, V(O6) and V′(O6), with populations of 2.64e and 2.64e, associated with the O6 oxygen lone pairs, and one V(C5,O6) disynaptic basin integrating for 2.39e which belongs to the C–O bonding region of acetone 3. The low population of the C–O bond together with the high population of the oxygen lone pairs indicates that the C–O bond of acetone 3 is very polarised.
Phase II, 2.80 Å ≥ d(C4–C5) > 2.33 Å and 3.00 Å ≥ d(C2–O6) > 2.62 Å, begins at P1. In this phase the most relevant changes imply the splitting of the V(N1,C2) disynaptic basin into two V(N1,C2) and V′(N1,C2) disynaptic basins with populations of 1.78e and 3.70e, and a slight decrease in the population of the V(C4) monosynaptic basin to 2.00e. The other basins maintain their populations as shown in phase I. On the acetone framework, the population of the V(C5,O6) disynaptic basin is 2.41e, while the two V(O6) and V′(O6) monosynaptic basins integrate for 2.65e and 2.70e, respectively.
Phase III, 2.34 Å ≥ d(C4–C5) > 2.14 Å and 2.62 Å ≥ d(C2–O6) > 2.46 Å, begins at P2. At this point, the V(N1,C2) and V′(N1,C2) disynaptic basins newly merge into one disynaptic basin V(N1,C2) integrating for 5.58e, while the population of the V(C4) monosynaptic basin has slightly decreased to 1.92e on the cis-IN moiety. The populations of the V(O6) and V′(O6) monosynaptic basins slightly increase to 2.71e and 2.77e for the acetone framework. These changes can be related to the GEDT that fluxes from cis-IN to acetone 3 during this polar reaction.
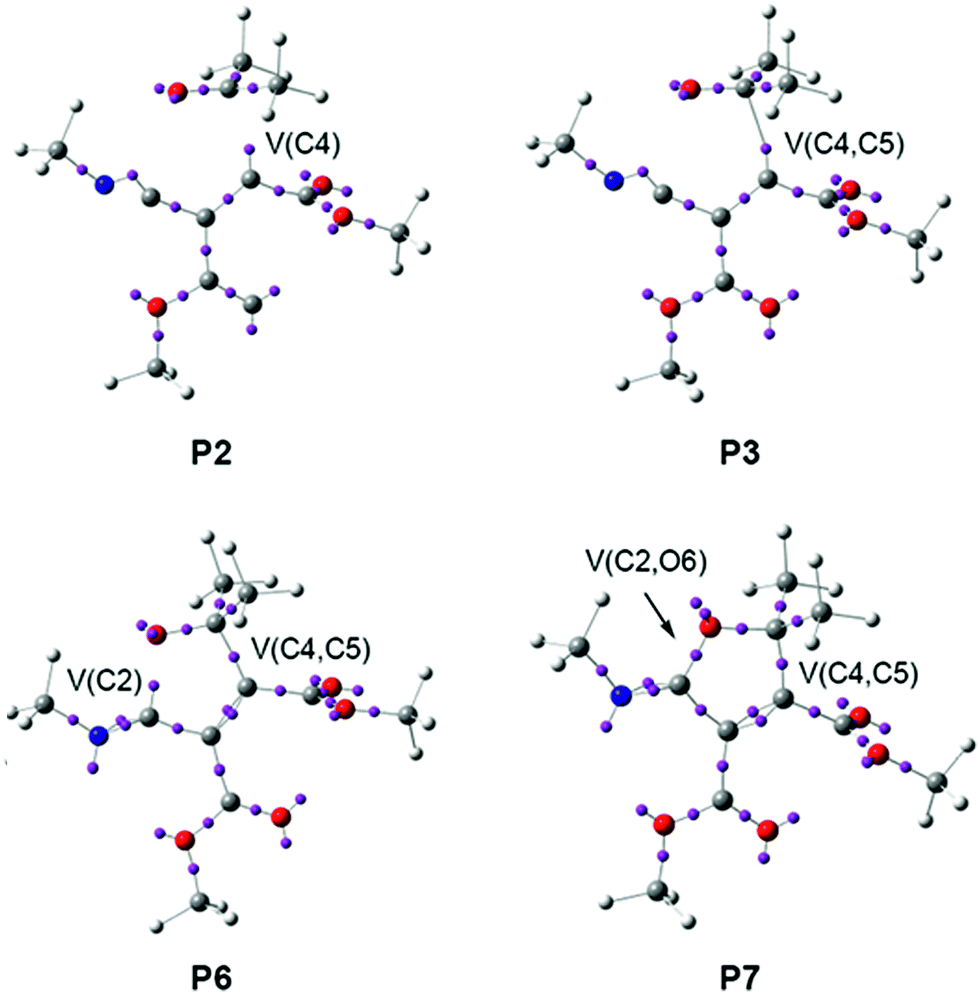
Phase IV, 2.14 Å ≥ d(C4–C5) > 2.07 Å and 2.46 Å ≥ d(C2–O6) > 2.41 Å, begins at P3, which corresponds to the TS of the reaction, TS2, d(C4–C5) = 2.141 Å and d(C2–O6) = 2.462 Å. At this point, the first most relevant change along the IRC is found; while the V(C4) monosynaptic basin present in cis-IN has disappeared, a new V(C4,C5) disynaptic basin, integrating for 1.87e, has appeared (see P2 and P3 in Fig. 6 and the change from V(C4), in green in P2, to V(C4,C5), in blue in P3, in Fig. 7). This change indicates that the formation of the new C4–C5 single bond has started at d(C4–C5) = 2.14 Å.
Phase V, 2.07 Å ≥ d(C4–C5) > 1.91 Å and 2.41 Å ≥ d(C2–O6) > 2.29 Å, starts at P4. After passing TS2, the most important change is the formation of a new V(N1) monosynaptic basin, which integrates for 0.80e, together with the depopulation of the V(N1,C2) disynaptic basin to 4.75e. At this point, the V(C3,C4) disynaptic basin present in cis-IN splits into two disynaptic basins, V(C3,C4) and V′(C3,C4), integrating for 1.68e and 1.61e, respectively. In addition, the population of the V(C4,C5) disynaptic basin slightly increases to 1.89e, and the populations of the V(O6) and V′(O6) monosynaptic basins of the acetone framework increase to 2.82e and 2.87e.
Phase VI, 1.91 Å ≥ d(C4–C5) > 1.65 Å and 2.29 Å ≥ d(C2–O6) > 2.14 Å, starts at P5. At this point, the V(N1,C2) disynaptic basin of the intermediate moiety newly splits into two V(N1,C2) and V′(N1,C2) disynaptic basins, which integrate for 2.14e and 2.09e, respectively, while the V(N1) monosynaptic basin reaches a population of 1.48e. On the acetone moiety, it may be seen that there is a depopulation of the V(C5,O6) disynaptic basin to 1.77e and a slight increase of the population of the V(O6) and V′(O6) monosynaptic basins to 2.94e and 2.96e.
The short phase VII, 1.65 Å ≥ d(C4–C5) > 1.58 Å and 1.95 Å ≥ d(C2–O6) > 1.75 Å, starts at P6. At this point, the second relevant topological change along the IRC takes place: a V(C2) monosynaptic basin is created at the C2 carbon, integrating for 0.20e (see the V(C2) monosynaptic basin in P6 in Fig. 6). This change shows the preparation of the intermediate fragment for the subsequent ring closure through the C–O bond formation. In this phase, the population of the V(C4,C5) disynaptic basin increases to 2.05e and the population of the V(N1) monosynaptic basin increases to 2.26e, whereas the populations of the V(C5,O6), V(N1,C2) and V′(N1,C2) disynaptic basins decrease to 1.53e, 1.83e and 1.84e, respectively, along with the progressing reaction.
Phase VIII, 1.58 Å ≥ d(C4–C5) ≥ 1.50 Å and 1.75 Å ≥ d(C2–O6) ≥ 1.37 Å, starts at P7 and ends at cycloadduct 4, d(C4–C5) = 1.502 Å and d(C2–O6) = 1.370 Å. At this point, the third most relevant change takes place with the formation of a new V(C2,O6) disynaptic basin integrating for 0.75e (see the V(C2,O6) disynaptic basin in P7 in Fig. 6 and the change from V(C2), in green in P6, to V(C2,O6), in blue in P7, in Fig. 7), while the populations of the V(O6) and V′(O6) monosynaptic basins have slightly decreased to 2.71e and 2.69e. This change indicates that the formation of the second C2–O6 single bond has started at a distance of 1.75 Å. At 2-iminofuran 4, the V(C2,O6) disynaptic basin reaches a population of 1.49e, while the V(C4,C5) disynaptic basin shows a population of 2.09e. The low population of the V(C2,O6) disynaptic basin indicates a very polarised C2–O6 single bond.
From the BET analysis of this domino reaction some appealing conclusions can be drawn: (i) during the nucleophilic attack of methyl isocyanide 1 on DMAD 2, the C2–C3 bond is formed at a distance of 1.90 Å through the donation of the electron-density of the carbene C2 lone pair to one of the two acetylenic carbons of DMAD 2; (ii) the formation of the new C2–C3 bond takes place with a high electron-density, 2.55e. Note that this value is higher than that associated with a C–C single bond; (iii) during the nucleophilic attack of isocyanide 1 on DMAD 2, a new V(C4) monosynaptic basin with an initial population of 0.52e appears at the C4 carbon as a consequence of the depopulation of the acetylenic C3–C4 bonding region. This V(C4) monosynaptic basin reaches a population of 2.06e at the intermediate cis-IN, while at the same time the C3–C4 bonding region is depopulated to reach 3.14e; (iv) during the nucleophilic attack of cis-IN on acetone 3, the formation of the C4–C5 single bond begins at a distance of 2.14 Å through the donation of the electron-density of the carbenoid C4 lone pair of cis-IN to the carbonyl C5 carbon of acetone 3; (v) formation of the second C2–O6 single bond takes place at the end of the cycloaddition path at a distance of 1.75 Å by sharing the electron-density of the V(C2) monosynaptic basin present at the C2 carbon and some of the electron-density of the monosynaptic basins associated with the oxygen O6 lone pairs; (vi) formation of the C2–O6 single bond begins after the complete formation of the C4–C5 bond, characterising the mechanism of the cycloaddition as a non-concerted two-stage one-step mechanism;36 and finally (vii) during the cycloaddition step, the N1C2 triple bond of cis-IN becomes a double bond characterised by the presence of two V(N1,C2) and V′(N1,C2) disynaptic basins at 2-iminofuran 4. At the same time, the C2 carbon is rehybridised from sp for cis-IN to sp2 for 2-iminofuran 4 as a consequence of the formation of the C2–O6 single bond. The orthogonal character of the new C2–O6 single bond with respect to the exocyclic N1–C2 double bond present in 2-iminofuran 4 indicates that the N1–C2 triple bond and C3–C4 double bond regions of the intermediate cis-IN do not participate directly in the cycloaddition reaction, as expected in a 1,3-dipole participating in a 1,3-dipolar cycloaddition.
Both analysis of the atomic movements of the cis-IN and acetone 3 molecules along the IRC associated with the cycloaddition step and the corresponding BET analysis indicate that the electron-density of the carbenoid C4 carbon and the carbonyl O6 oxygen lone pairs mainly participate in the formation of the C4–C5 and C2–O6 single bonds during the reaction; consequently, this cycloaddition should be classified as [2n + 2n], in which two lone pairs are involved, and not as a [4π + 2π] 1,3-dipolar cycloaddition.
(iv) What is the electronic structure of intermediate cis-IN and the origin of the high reactivity of carbonyl compounds towards this intermediate?
At first, the intermediate cis-IN can be represented by either of the Lewis resonant structures A and B given in Scheme 5. Both structures would represent a zwitterionic intermediate in which the negative charge is located at the C4 carbon atom, while the positive charge can be located at the C2 or N1 atoms belonging to the methyl isocyanide framework. Experimental chemists represent cis-IN by means of the 1,3-zwitterionic structure A, since it justifies the participation of cis-IN in a [3 + 2] cycloaddition towards carbonyl derivatives.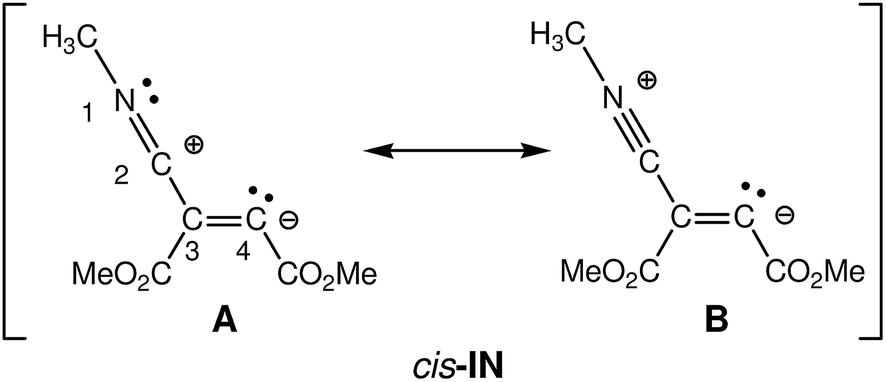 | ||
| Scheme 5 Lewis structures representing zwitterionic intermediate cis-IN. | ||
NPA and ELF analysis of the electronic structure of the intermediate cis-IN yield different representations for this intermediate. NPA of cis-IN clearly shows the zwitterionic character of this intermediate: while the methyl isocyanide framework is positively charged, the DMAD framework is negatively charged (see Fig. 8a). However, while NPA indicates that the positive charge is mainly located at the C2 carbon, in agreement with Lewis structure A, the results also indicate that the negative charge is mainly located at the C3 carbon and the carboxylate oxygen atoms, the C4 carbon having a negligible negative charge of −0.07e.
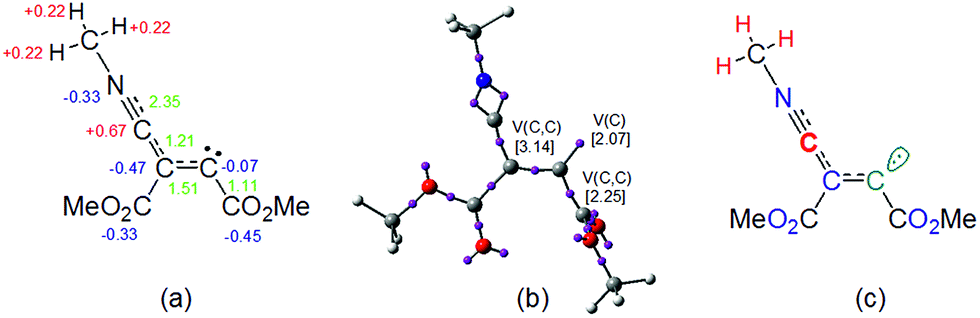 | ||
| Fig. 8 Electronic representation of the structure of carbenoid intermediate cis-IN based on (a) NPA, (b) ELF analysis and (c) the mixture of both analyses. Green values in (a) indicate the bond order of the corresponding bond. | ||
On the other hand, the ELF topology of cis-IN shows the presence of a monosynaptic basin at C4, V(C4), integrating for 2.07e (see Fig. 8b). In addition, the C3–C4 double bond regions of the Lewis structures A and B have a noticeable depopulation, 3.14e. This behaviour can account for the negligible negative charge found at the C4 carbon, −0.07e and thus both Lewis structures, A and B, can be ruled out as representations for the intermediate cis-IN.
Consequently, in spite of the zwitterionic character of the intermediate cis-IN, it appears that the reactivity of this intermediate towards carbonyl compounds cannot be related to its zwitterionic character but rather to the singlet carbenoid character of the C4 carbon that grants a high nucleophilic character to this molecule (see the C4 carbon in green in Fig. 8c).
In order to explain the high reactivity of acetone 3 towards this intermediate, an analysis of the molecular electrostatic potential (MEP) at cis-IN and the corresponding TS2 was also performed (see Fig. 9). The MEP of cis-IN allows two appealing conclusions to be reached: (i) around the nucleophilic C4 carbon a low negative electrostatic potential (in red) is found (see Fig. 9a). Note that the carboxylate oxygen atoms show the highest values. This behaviour is in agreement with the unappreciable negative charge found at the C4 atom; (ii) interestingly, the region of MEP with the highest positive value of cis-IN corresponds to the methyl substituent present in the isocyanide framework.
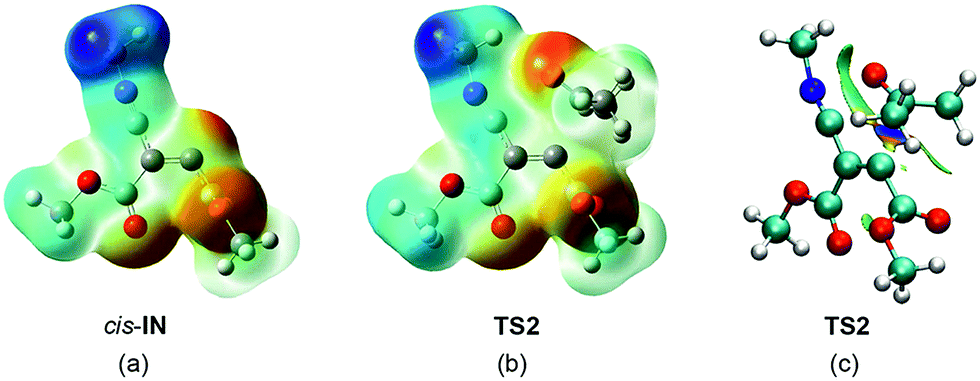 | ||
| Fig. 9 The MEP of carbenoid intermediate cis-IN (a) and TS2 (b), and NCIs for TS2 (c). | ||
The MEP of the carbenoid intermediate cis-IN shows the special characteristic of this intermediate that favours the nucleophilic attack on carbonyl derivatives. As can be seen in Fig. 9, the analysis of the MEP of TS2 clearly shows that during the nucleophilic attack of cis-IN on acetone 3, the GEDT that takes place in this polar process gives rise to an increase of electron-density of the oxygen carbonyl atom. This feature, which is unfavourable in an uncatalysed nucleophilic addition to carbonyl compounds, is favoured in TS2 by the presence of the positively charged methyl group that electrostatically stabilises the negative charge developed at the carbonyl oxygen atom (see Fig. 9b). Note that during the formation of the first C4–C5 single bond, the GEDT that takes place from cis-IN to acetone 3, 0.40e at P5, mainly locates the electron-density at the carbonyl oxygen atom (see the integration of the V(O6) and V′(O6) monosynaptic basins at P5 in Table 5).
Finally, in order to rule out a stabilisation of TS2 by a hydrogen bond between the carbonyl oxygen atom and one hydrogen atom of the methyl group, an analysis of the NCI at TS2 was performed. As can be seen in Fig. 9c, although some weak interactions between the carbonyl oxygen atom and the methyl isocyanide framework appear (in green), no hydrogen bond interaction between the carbonyl oxygen and the hydrogen of the methyl group is observed. Note that strong hydrogen bonds appear as a dark turquoise surface. Indeed, the NCI between the carbonyl O6 oxygen atom and the C2 carbon of the isocyanide framework is stronger than that involving the hydrogen of the methyl group, indicating the favourable interaction preceding the C2–O6 single bond formation.
Consequently, we can conclude that the high reactivity of the intermediate cis-IN towards carbonyl derivatives is due to two specific features: (i) the carbenoid character of the sp2 hybridised C4 carbon rather than the negative charge on a carbanionic center and, (ii) the special geometric disposition of the alkyl substituents in isocyanides that electrostatically favours the GEDT during the nucleophilic attack of these carbenoid intermediates on carbonyl compounds.
Conclusions
The high reactivity of acetone 3 towards the nucleophilic carbenoid intermediate cis-IN, generated in situ by the addition of methyl isocyanide 1 to DMAD 2, has been studied using DFT methods at the MPWB1K/6-311G(d,p) computational level through the combination of the exploration and characterisation of the PESs associated with this MC reaction and analysis based on the MEDT, consisting of the analysis of the reactivity indices derived from the conceptual DFT at the ground state of the reagents and the BET study for the corresponding reaction paths.This MC reaction is a domino process that comprises two consecutive reactions: (i) formation of the carbenoid intermediate trans-IN, which quickly equilibrates with the thermodynamically more stable cis-IN; and (ii) the nucleophilic capture of acetone 3 by the carbenoid intermediate cis-IN yielding the formation of the final 2-iminofuran 4.
Analysis of the relative Gibbs free energies in acetonitrile indicates that while the initial nucleophilic attack of the carbene methyl isocyanide 1 on DMAD 2 is the RDS of this MC reaction, once the intermediate cis-IN is formed, it quickly and irreversibly captures acetone 3.
Analysis of the DFT reactivity indices for the intermediate cis-IN clearly accounts for its high nucleophilic character, entirely at the carbenoid C4 carbon. These behaviours explain the high reactivity of the intermediate cis-IN in polar reactions towards electron-deficient carbonyl compounds.
BET analysis of the two reactions involved in this domino process makes it possible to draw some appealing conclusions concerning the bonding changes occurring in this MC reaction: (i) during the nucleophilic attack of isocyanide 1 on DMAD 2, the C2–C3 bond is formed at a distance of 1.90 Å through the donation of the electron-density of the carbene C2 lone pair to one of the two acetylenic carbons of DMAD. Formation of this C2–C3 bond takes place with a high electron-density, 2.55e; (ii) during the nucleophilic attack of isocyanide 1 on DMAD 2, a new V(C4) monosynaptic basin with an initial population of 0.52e appears at the C4 carbon as a consequence of the depopulation of the acetylenic C3–C4 triple bond. This V(C4) monosynaptic basin reaches a population of 2.06e for the intermediate cis-IN; (iii) during the nucleophilic attack of cis-IN on acetone 3, the formation of the C4–C5 single bond begins at a distance of 2.14 Å through the donation of the electron-density of the carbenoid C4 lone pair of cis-IN to the carbonyl C5 carbon of acetone 3; (iv) formation of the second C2–O6 single bond takes place at the end of the cycloaddition path at a distance of 1.75 Å by sharing the electron-density of the V(C2) monosynaptic basin present at the C2 carbon and some electron-density of the monosynaptic basins associated with the O6 oxygen lone pairs; and (v) formation of the C2–O6 single bond begins after the complete formation of the C4–C5 bond. This behaviour characterises the mechanism of the cycloaddition as a non-concerted two-stage one-step mechanism.36
An analysis of the electronic structure of the intermediate cis-IN makes it possible to explain the high reactivity of this intermediate towards carbonyl derivatives. Two specific features of this intermediate enable this MC reaction: (i) the carbenoid character of the sp2 hybridised C4 carbon of cis-IN, rather than the negatively charged carbanionic center as it is represented in the bibliography; and, (ii) the special geometric disposition of the alkyl substituent present in the isocyanide, which electronically stabilises the negative charge gathered at the carbonyl oxygen atom during the nucleophilic attack.
The present MEDT study establishes that the high nucleophilic character of the carbenoid intermediate cis-IN together with the specific approach mode of the carbonyl CO double bond during the nucleophilic attack of the sp2 hybridised carbenoid center of cis-IN on the carbonyl carbon of acetone 3 make the formation of the C–C single bond with a very low activation enthalpy, 3.3 kcal mol−1, possible, without any external electrophilic activation of the carbonyl group, while the geometric and electronic features of this intermediate favour the subsequent ring closure through the downhill formation of the C–O single bond, thus providing the answers to the three unresolved questions posed concerning the electronic structure of the intermediate involved and the molecular mechanism of these experimentally widely investigated MC reactions.
Finally, both the analysis of the atomic movements of the molecules cis-IN and acetone 3 along the IRC associated with the cycloaddition step and the corresponding BET analysis allow characterisation of the mechanism of this reaction as a [2n + 2n] cycloaddition in which two lone pairs are involved in the formation of the new C4–C5 and C2–O6 single bonds. These findings make it possible to reject the 1,3-dipolar cycloaddition mechanism for the cycloaddition reactions of carbonyl compounds to these nucleophilic carbenoid intermediates.
Acknowledgements
This work has been supported by the Ministerio de Economía y Competitividad of the Spanish Government, project CTQ2013-45646-P, by FONDECYT through Project 1140341, by the Millennium Nucleus of Chemical Processes and Catalysis (CPC), grant number NC120082 and by DI-UNAB-793-15/R. M. R.-G. thanks the Ministerio de Economía y Competitividad for a pre-doctoral contract co-financed by the European Social Fund (BES-2014-068258).References
-
(a) A. Domling, Chem. Rev., 2006, 106, 17 CrossRef PubMed
; (b) F. Millich, Chem. Rev., 1972, 2, 101 CrossRef
-
(a) V. Nair, A. U. Vinoda, N. Abhilasha, R. S. Menona, V. Santhia, R. L. Varmaa, S. Vijia, S. Mathewa and R. Srinivasb, Tetrahedron, 2003, 59, 10279 CrossRef CAS PubMed
- V. Nair and A. U. Vinoda, Chem. Commun., 2000, 1019 RSC
-
(a) A. A. Esmaeili and M. Darbanian, Tetrahedron, 2003, 59, 5545 CrossRef
- A. Shaabani, A. H. Rezayan, S. Ghasemi and A. A. Sarvary, Tetrahedron Lett., 2009, 50, 1456 CrossRef CAS PubMed
- I. Yavari and H. Djahaniani, Tetrahedron Lett., 2005, 46, 7491 CrossRef CAS PubMed
- L.-L. Zhao, S.-Y. Wang, X.-P. Xu and S.-J. Ji, Chem. Commun., 2013, 49, 2569 RSC
-
(a) F. M. Dean, in Advances in Heterocyclic Chemistry, ed. A. R. Katritzky, Academic, New York, 1982, vol. 30, p. 167 Search PubMed
- W. P. Pei, J. Pei, S. H. Li and X. L. Ye, Synthesis, 2000, 2069 CrossRef CAS
-
(a) P. Geerlings, F. de Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS PubMed
- X. Krokidis, S. Noury and B. Silvi, J. Phys. Chem. A, 1997, 101, 7277 CrossRef CAS
-
(a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS
-
(a) C. E. Check and T. M. Gilbert, J. Org. Chem., 2005, 70, 9828 CrossRef CAS PubMed
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908 CrossRef CAS
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed
-
(a) H. B. Schlegel, J. Comput. Chem., 1982, 2, 214 CrossRef PubMed
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS
-
(a) C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef
-
(a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS
-
(a) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS PubMed
-
(a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS PubMed
-
(a) R. G. Parr and W. Yang, Annu. Rev. Phys. Chem., 1995, 46, 701 CrossRef CAS PubMed
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922 CrossRef CAS
-
(a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS
-
(a) L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615 CrossRef CAS PubMed
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133 CrossRef
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486 RSC
-
(a) S. Berski, J. Andrés, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2003, 107, 6014 CrossRef CAS
-
(a) R. Thom, Structural Stability and Morphogenesis: An Outline of a General Theory of Models, Addison-Wesley Publishing Company, Inc., Reading, MA, 1976 Search PubMed
-
(a) X. Krokidis, V. Goncalves, A. Savin and B. Silvi, J. Phys. Chem. A, 1998, 102, 5065 CrossRef CAS
-
(a) A. Savin, A. D. Becke, J. Flad, R. Nesper, H. Preuss and H. G. Vonschnering, Angew. Chem., Int. Ed., 1991, 30, 409 CrossRef PubMed
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597 CrossRef CAS
-
(a) E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, J. Cohen and A. W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian Inc, Wallingford, CT, 2009 Search PubMed
- L. R. Domingo, J. A. Saéz, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73, 8791 CrossRef CAS PubMed
- L. R. Domingo, RSC Adv., 2014, 4, 32415 RSC
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417 CrossRef CAS
- P. Jaramillo, L. R. Domingo, E. Chamorro and P. Pérez, J. Mol. Struct.: THEOCHEM, 2008, 865, 68 CrossRef CAS PubMed
- A. Savin, J. Chem. Sci., 2005, 117, 473 CrossRef CAS
- B. Silvi, J. Mol. Struct., 2002, 614, 3 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2015 |