
New insights into electrolyte-component biased and transfer- and transport-limited charge recombination in dye-sensitized solar cells
Abstract
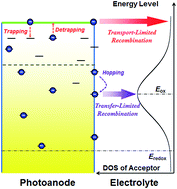
We carried out a time-resolved charge extraction (TRCE) study on the charge recombination dynamics of open-circuit dye-sensitized solar cells (DSSCs) by examining the temporal evolution of electron density with varying the concentration of the electrolyte component tert-butylpyridine (TBP). The charge recombination dynamics extracted from TRCE results exhibit distinctly different temporal behavior depending on the photovoltage or electron density. We proposed a theoretical model of electron density dependent charge recombination, which unifies the transport- and transfer-limited charge recombination mechanisms. This model, as rationalized by transient photovoltage (TPV) kinetics, can account well for the TRCE results. The relevance between electron transport and charge transfer in the recombination process was unraveled, and the electron density dependent pathway of charge recombination was elucidated.
 Please wait while we load your content...
Please wait while we load your content...

