
Super long-life supercapacitor electrode materials based on hierarchical porous hollow carbon microcapsules†
Abstract
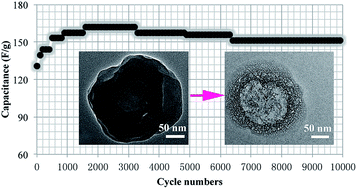
Remarkable supercapacitor electrodes with a high specific supercapacitance and a super long cycle life were achieved by using hierarchical porous hollow carbon microcapsules (HPHCMs) as active materials. HPHCMs were prepared by a facile chemical route based on pyrolysis of a soft sacrificial template involving a non-crosslinked core of poly(styrene-r-methylacrylic acid) and a crosslinked shell of poly(styrene-r-divinylbenzene-r-methylacrylic acid), which were synthesized by using traditional radical polymerization and emulsion polymerization. The results of scanning electron microscopy, transmission electron microscopy and Brunauer–Emmett–Teller characterizations revealed that HPHCM possessed the desired pore structure with apparent macro-/meso- and micropores, which not only provided a continuous electron-transfer pathway to ensure good electrical contact, but also facilitated ion transport by shortening diffusion pathways. As electrode materials for supercapacitor, a high specific capacitance of 278.0 F g−1 was obtained at the current density of 5 mA cm−2. Importantly, after 5000 potential cycles in 2 M KOH electrolyte at the discharge current density of 20 mA cm−2, the capacitance actually increased from 125 to 160 F g−1 and then remained 151 F g−1, corresponding to a capacitance retention of 120%, likely due to electrochemical self-activation.
 Please wait while we load your content...
Please wait while we load your content...

