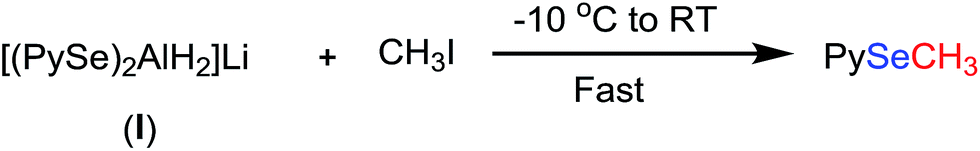DOI:
10.1039/C5RA15577K
(Paper)
RSC Adv., 2015,
5, 78669-78676
A mechanistic study of the synthesis, single crystal X-ray data and anticarcinogenic potential of bis(2-pyridyl)selenides and -diselenides†
Received
4th August 2015
, Accepted 8th September 2015
First published on 9th September 2015
Abstract
The reaction of bis(organyl)diselenide with a reducing agent, such as LiAlH4, NaBH4, Li(C2H5)3BH, etc., generally leads to cleavage of the Se–Se bond resulting in the formation of the corresponding organylselenols/selenolates. However, this work for the first time demonstrates the scisson of the C(pyridine)–Se bond in bis(2-pyridyl)diselenides with LiAlH4. The reaction affords analytically pure bis(2-pyridyl)selenides in near quantitative yields. The reaction pathway involves the formation of a selenated aluminato complex followed by the scission of the C(pyridine)–Se bond and generation of LiAlSeH2. The generation of LiAlSeH2 was established by experimental and NMR analysis. The mechanism of the reaction has been supported by theoretical analysis. Single crystal X-ray structure determination of bis(3-methyl-2-pyridyl)selenide (2e) was performed and it shows that the molecules are self-assembled in a 2D-network of C–H⋯N hydrogen bonds and π⋯π stacking interactions. The synthesized compounds were also evaluated against the Raji cancer cell line (acute lymphoid leukemia).
Introduction
The recent surge in the field of selenium chemistry stems from the realization of organoselenides as materials and biologically significant molecules.1–3 A number of reagents such as sodium borohydride,4 hydrazine hydrate,5 alkyllithiums/lithium dialkylamides,6 Grignard reagents,7 etc. have been utilized to incorporate selenium into a variety of organyl moieties.8–10 Lithium aluminum hydride (LiAlH4) has been rarely used in these reactions, which in part is due to difficulty in its handling especially, at ambient conditions. Ishihara et al.11 exploited the potential of LiAlH4 and synthesized a variety of organoselenium compounds. The method involved the reaction of elemental selenium with LiAlH4 leading to the generation of LiAlSeH2. LiAlH4 has been recently used by our group for the cleavage of Se–Se bond in bis(2-pyridyl)diselenides12a and bis(3-(4-chloro-N,N-tetraisopropylpyridine)-2,6-dicarboxamide)diselenide.12b
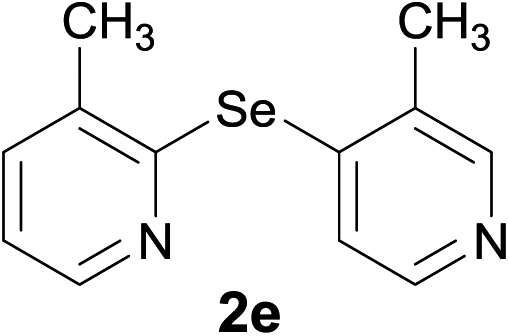
Most of the studies concerning pyridylselenides focuses on the synthesis of bis(2-pyridyl)diselenides. However, the corresponding chemistry of bis(2-pyridyl)selenides is underexplored although they have shown significant utility as biologically significant molecules.13 This is mainly due to the non-availability of a suitable methodology for the synthesis of these compounds. Bis(2-pyridyl)selenides have been largely obtained as a by-product in the synthesis of the corresponding diselenides.4c There are only a few reports on the synthesis of bis(2-pyridyl)selenides. Bhasin et al.14 used the metal-halogen exchange pathway involving 5-bromo-2-iodopyridine and selenium dichloride to synthesize the corresponding monoselenides. In continuation of our investigation on organoselenium compounds,6,12,15 we report here a mechanistic study on the reaction involving LiAlH4 and bis(2-pyridyl)diselenides leading to the generation of analytically pure bis(2-pyridyl)selenides. The reaction occurs through the scission of C(pyridine)–Se bond in bis(2-pyridyl)diselenides. To the best of our knowledge, there is no study that reports the scission of the C–Se bond in bis(organyl)diselenide. The single crystal X-ray structure of bis(3-methyl-2-pyridyl)selenide (2e) has been determined and it was observed that the molecules are self-assembled in a 2D-network of C–H⋯N hydrogen bonds and π⋯π stacking interactions. The synthesized compounds were evaluated as anticarcinogenic agents against the Raji cancer cell line (acute lymphoid leukemia), using the MTT colorimetric assay.
Results and discussion
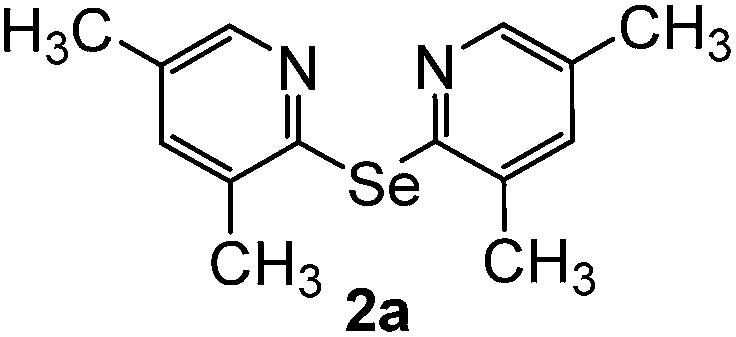
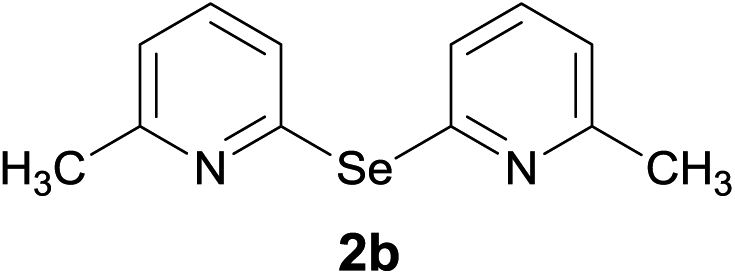
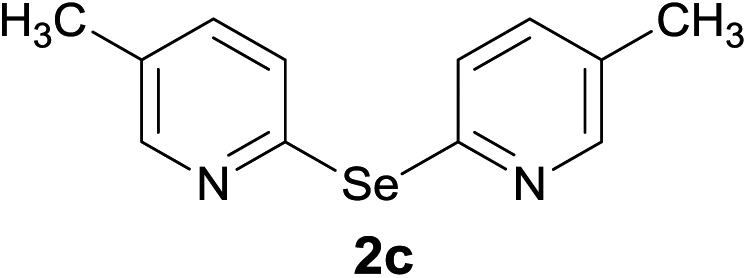
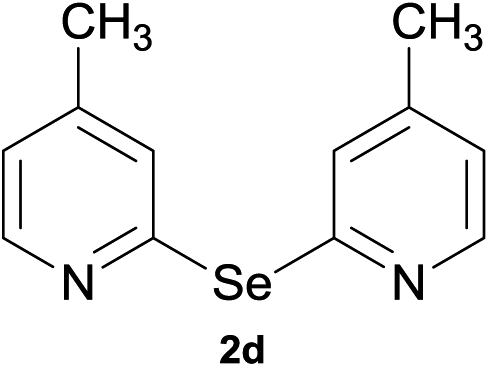
Bis(3,5-dimethyl-2-pyridyl)diselenide (1a) was reacted with LiAlH4 in THF at −10 °C. The reaction mixture was slowly brought to the room temperature and refluxed for 16 h. The progress of the reaction was monitored by TLC and GC-MS analysis. The reaction was stopped when all of the diselenide was completely consumed. After the work-up and purification procedures, bis(3,5-dimethyl-2-pyridyl)selenide (2a) was obtained in an excellent yield (entry 1, Table 1). Following the same procedure, other bis(2-pyridyl)selenides (2b–2f) were also synthesized (entries 2–6, Table 1).
Table 1 Synthesis of bis(2-pyridyl)selenides by using LiAlH4
In order to check the influence of the solvent on the rate of the reaction, these reactions were also carried out in DMF. Contrary to the expectation, the reactions in DMF were almost two times slower than in THF. In addition, the yields of the reactions were lower than in THF (Table 1). The comparative lower yields in DMF could be due to the competing reaction of LiAlH4 with DMF. LiAlH4 has the tendency to react with alkyl/arylamides to give the corresponding amines.
Mechanistic study
The exclusive formation of monoselenides suggests that one of the two C(pyridine)–Se bonds in bis(2-pyridyl)diselenides is selectively cleaved in the presence of LiAlH4. This is contrary to the generally accepted exposition that the treatment of a reducing agent, such as LiAlH4,12 NaBH4,4 Li(C2H5)3BH,16 etc., with bis(organyl)diselenide cleaves the Se–Se bond and generate the corresponding organylselenols/selenolates only. It is pertinent to mention that there is no report that mentions the scission of the C–Se bond in bis(organyl)diselenide.
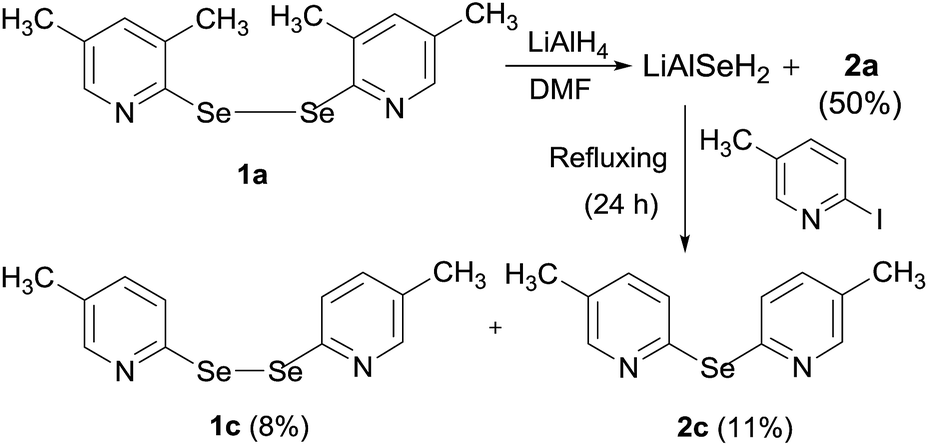
It is further postulated that LiAlH4 retains the cleaved selenium atom resulting in the formation of LiAlSeH2. To confirm the formation of LiAlSeH2, we added 2-iodo-5-methylpyridine to a mixture of (1a) and LiAlH4. Our thinking was that 2-iodo-5-methylpyridine would react with the in situ formed LiAlSeH2 and afford the corresponding mono- or diselenide. This was actually the case as bis(5-methyl-2-pyridyl)diselenide (1c) and bis(5-methyl-2-pyridyl)selenide (2c) along with 2a were obtained from the reaction mixture (Scheme 1). The lower yield of the products is mainly due to the difficulty in the purification of the crude product by column chromatography. Since these products travelled very closely in the chromatographic column, the chromatography procedure was performed three times to get analytically pure products.
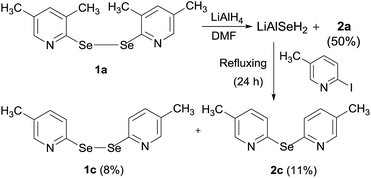 |
| | Scheme 1 Reaction sequence exhibiting the generation of LiAlSeH2. | |
The generation of LiAlSeH2 was also confirmed by the NMR analysis. A mixture of (1c) and LiAlH4 was refluxed in THF for 15 h. The solvent from the reaction mixture was removed and a solid greyish powder was obtained. The 1H NMR spectrum of the powder was taken in CDCl3 that showed two prominent peaks at δ 1.74 and 3.62 ppm (Fig. 1). These chemical shift values are close to those reported by Ishihara et al. for LiAlSeH2.11 The latter two observations confirms the formation of LiAlSeH2 during the reaction of LiAlH4 with bis(2-pyridyl)diselenides.
 |
| | Fig. 1 1H NMR spectrum of in situ generated LiAlSeH2. | |
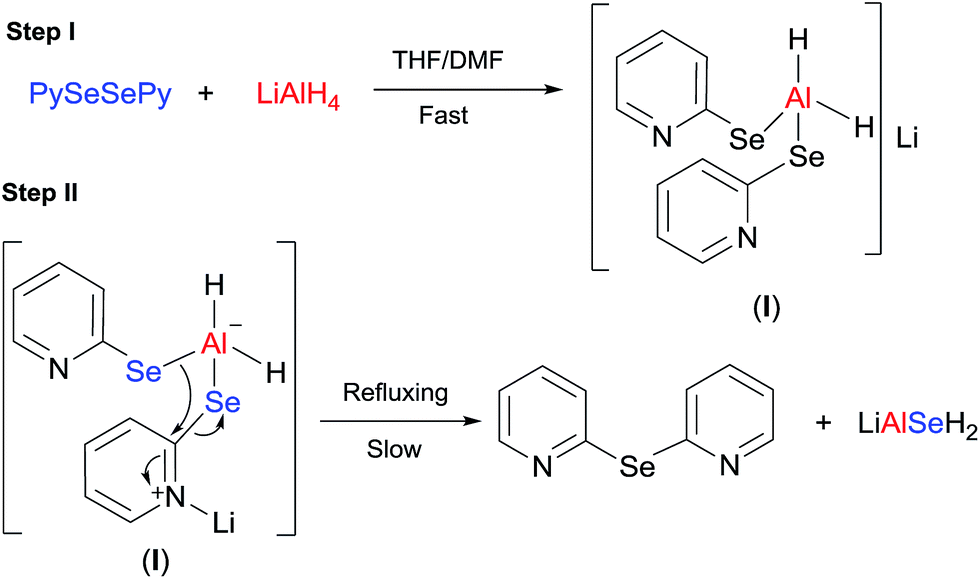
From the aforementioned facts, the following mechanistic steps have been proposed. The first step is a fast reaction involving the cleavage of the Se–Se bond and simultaneous formation of a selenated aluminato complex (I) (Scheme 2). Lithium is attached to the one of two pyridine rings (from the optimized structure, Fig. 2, discussed later) and this ring behaves as an electrophile and initiates the reaction involved in the second step. The second step is a slow and energy intensive process that involves the transfer of one of the two pyridyl moieties, attached to the complex I, to the adjacent pySe unit as shown in Scheme 2 (pyridine-shift reaction). This generates bis(2-pyridyl)selenide and LiAlSeH2 as the final products.
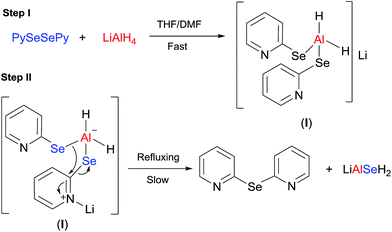 |
| | Scheme 2 Mechanism of the reaction. | |
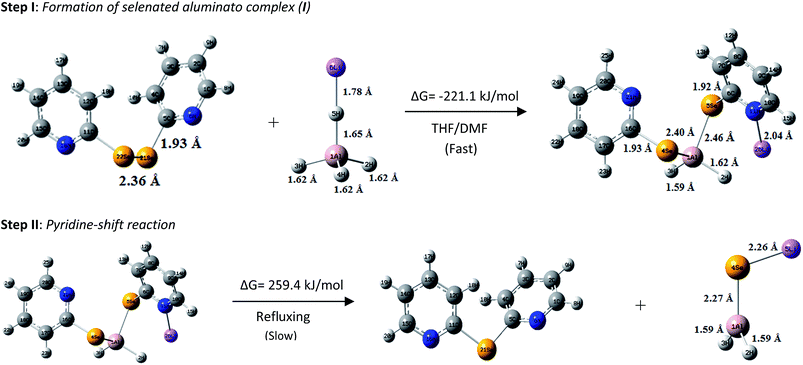 |
| | Fig. 2 Reaction mechanism with optimized structures of reactants, products and intermediate species. | |
The mechanism was tested by investigating a number of reactions. Bis(2-pyridyl)diselenide was refluxed in DMF/THF for 48 h but there was no indication of the formation of the corresponding monoselenide. This indicates that the diselenide is perfectly stable at elevated temperatures and it is LiAlH4 that aides in the formation of the monoselenide. Further, the reaction of LiAlH4 and bis(2-pyridyl)selenide in DMF and THF was investigated. In spite of refluxing the reaction mixture for more than 48 h, no interaction between LiAlH4 and bis(2-pyridyl)selenide was observed. This establishes that the cleavage of Se–Se bond with LiAlH4 precedes the formation of the aluminato complex (I) and subsequently the pyridine-shift reaction. In another variation, we examined the reaction of bis(2-pyridyl)diselenide with NaBH4 in DMF. The solution containing the diselenide and NaBH4 was refluxed for several hours; however there was no indication of the formation of the monoselenide. All these observations suggests an unique ability of LiAlH4 in inducing the cleavage of C–Se bond in bis(2-pyridyl)diselenides.
In our earlier studies,12 we have reported the synthesis of unsymmetrical selenides from the reaction of diselenides with LiAlH4 followed by treatment with a strong electrophile such as iodomethane. We revisited this chemistry in order to investigate if any symmetrical monoselenide is formed in this reaction also. Bis(2-pyridyl)diselenide was reacted with LIAlH4 in THF at −10 °C. The resulting reaction mixture was treated with iodomethane and refluxed for 36 h. During this time the reaction was continuously monitored by GC-MS. There was no trace of bis(2-pyridyl)selenide at any time of the reaction. The whole of bis(2-pyridyl)diselenide was converted into (2-methylselenenyl)pyridine (Scheme 3). It appears that the attack of CH3I on the complex, I, is faster and more favourable than the pyridine-shift reaction.
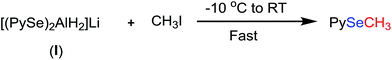 |
| | Scheme 3 Reaction of aluminato complex (I) with iodomethane. | |
Theoretical validation of the mechanism
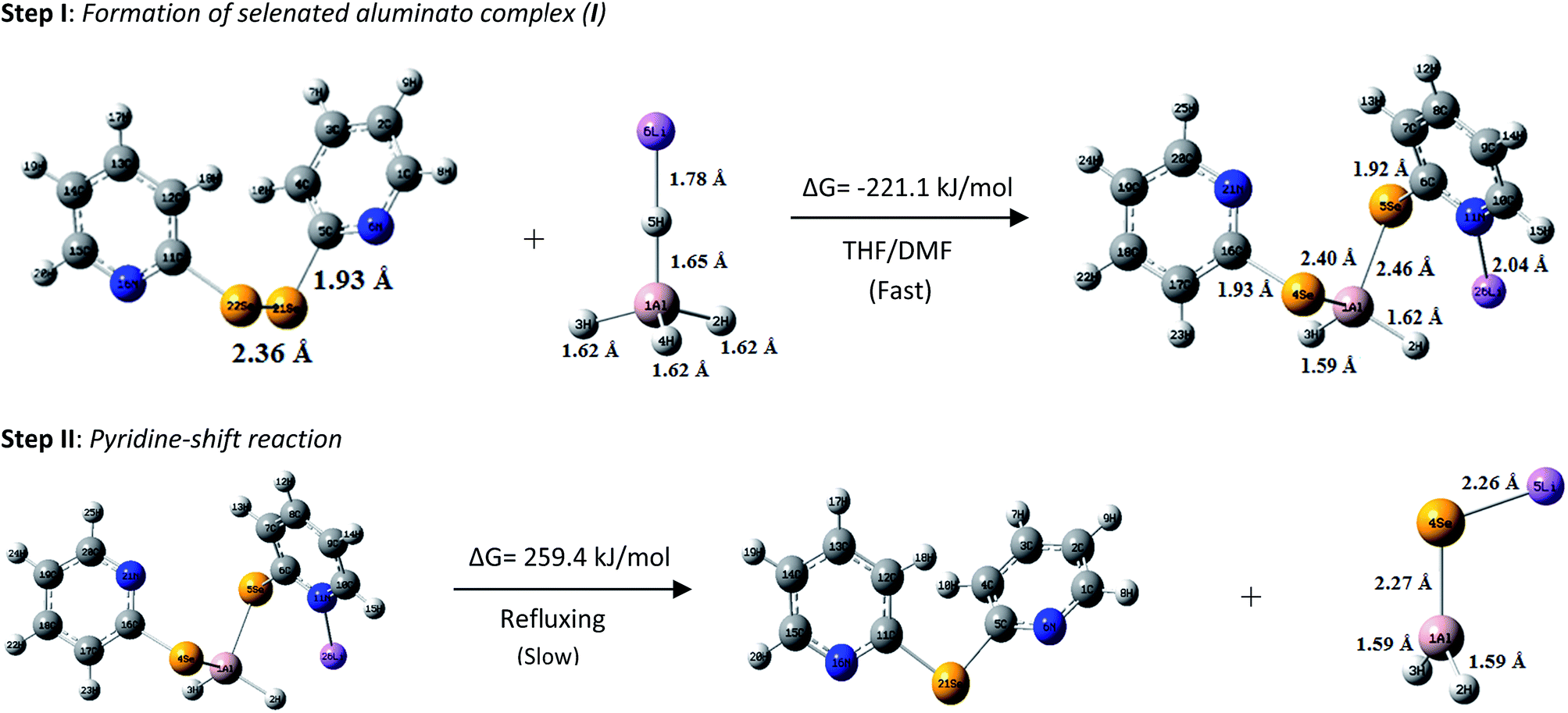
Gibbs free energy was calculated by using the closed-shell Becke–Lee–Yang–Parr hybrid exchange–correlation three-parameter functional (B3LYP) in combination with 6-31 + G(d,p) basis set in THF solvent to theoretically support the reaction mechanism shown in Scheme 2. The reaction shown in step I involves Gibbs free energy change (ΔG) of −221.1 kJ mol−1 which strongly provides evidence of a spontaneous reaction involving the formation of the aluminato complex (I) (Fig. 2). The second step involves a positive ΔG value of 259.4 kJ mol−1, which indicates an energy intensive process. This actually is the case as the second step requires refluxing for a long duration of time. The optimized structures of 1f, LiAlH4, aluminato complex and LiAlSeH2 are given in Fig. 2. The optimized structures of 1f and LiAlH4 agree well with the corresponding experimentally determined structures.6c,17 The Al–H bond distances are shorter in LiAlSeH2 than in LiAlH4. Lithium is attached to the Se atom in LiAlSeH2, whereas it is bonded to one of the four hydrogen atoms in LiAlH4.
Synthesis of monoselenides directly from elemental selenium and LiAlH4
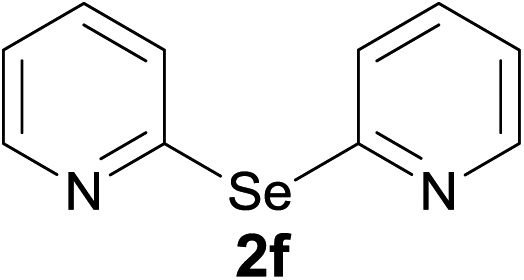
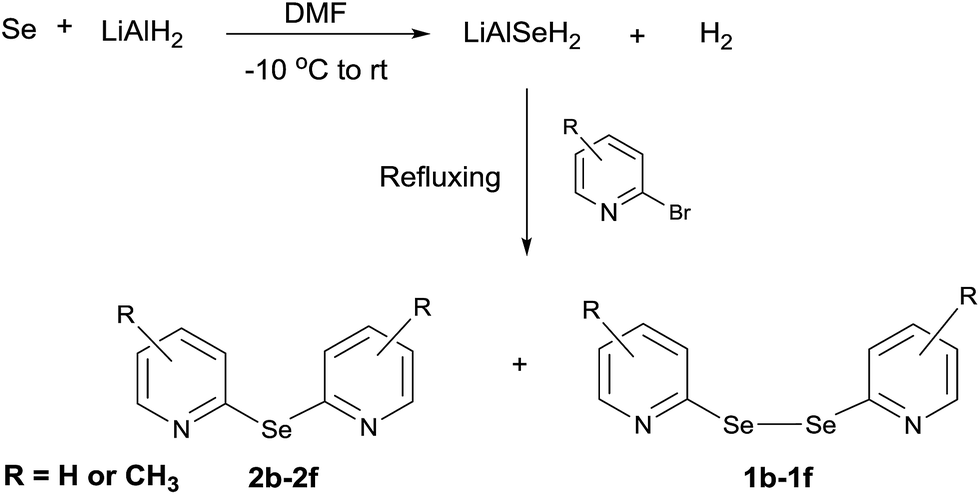
Next, we investigated the direct formation of monoselenides from the reaction of LiAlH4 and elemental selenium. LiAlH4 (1.0 equiv.) was added to a suspension of elemental selenium in dry DMF. A grayish suspension indicating the formation of LiAlSeH2 was obtained. Addition of 2-bromopyridine (1.0 equiv.) to this suspension and 3 h refluxing gave bis(2-pyridyl)selenide (2f) and bis(2-pyridyl)diselenide (1f) in 72% and 23% yields, respectively (Scheme 4).
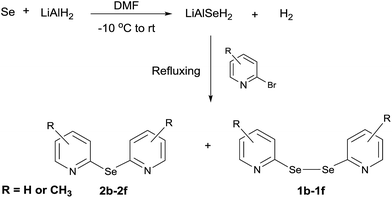 |
| | Scheme 4 Synthesis of bis(2-pyridyl)selenides and -diselenides. | |
Following the same procedure, other bis(2-pyridyl)selenides (2b–2e) and bis(2-pyridyl)diselenides (1b–1e) were also obtained (Scheme 4 and Table 2). Interestingly, in all these reactions the monoselenide was the major product formed even when the proportion of 2-bromopyridine was increased from 1.0 equiv. to 2.0 equiv. With the other reported methodologies,5a,12,18 the diselenide is always the major product formed. It appears that this methodology is more suitable for the synthesis of bis(2-pyridyl)selenides instead of bis(2-pyridyl)diselenides. Part of this appears to come from the scission of C–Se bond in bis(2-pyridyl)diselenide.
Table 2 Synthesis of mono- and diselenides of pyridine
| Entry |
Starting material |
Products |
| Monoselenide |
Diselenide |
| 1 |
2-Bromo-6-methylpyridine |
2b (69%) |
1b (24%) |
| 2 |
2-Bromo-5-methylpyridine |
2c (71%) |
1c (22%) |
| 3 |
2-Bromo-4-methylpyridine |
2d (70%) |
1d (27%) |
| 4 |
2-Bromo-3-methylpyridine |
2e (70%) |
1e (26%) |
| 5 |
2-Bromopyridine |
2f (72%) |
1f (23%) |
Single crystal X-ray structure determination of 2e
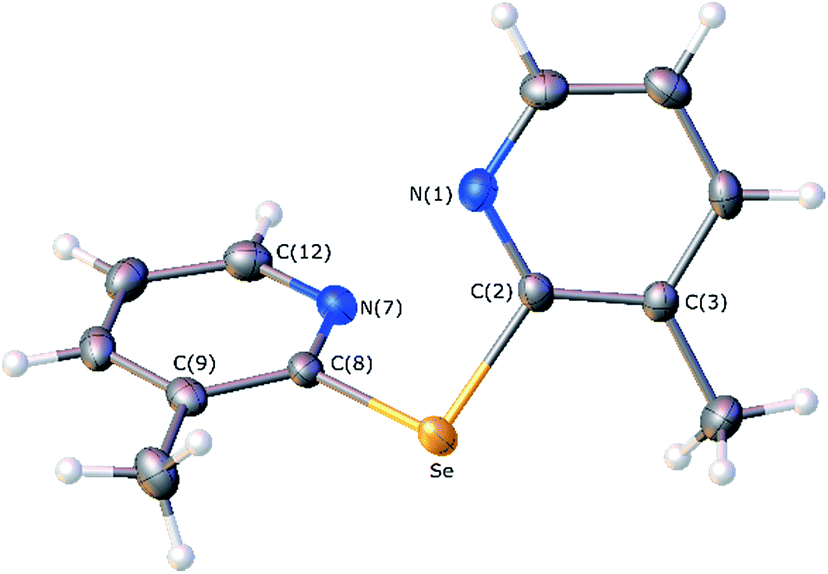
The crystals of 2e suitable for X-ray diffraction analysis were grown by a slow evaporation of a saturated solution of hexane and ethyl acetate. A thermal ellipsoid drawing of 2e, presented in Fig. 3, shows that the molecule adopts a twisted conformation, in which the nitrogen atoms of two pyridine rings face each other with a dihedral angle between the two aromatic rings of 66.69(7)°. The selected bond lengths involving the selenium centre, gathered in Table 3, are in close agreement with those determined for bis(2-pyridyl)selenide analogue 2f.19 However, in this compound a different conformation is adopted, which is characterized by the opposite spatial disposition of the pyridine nitrogen relatively to the C–Se–C central fragment and a dihedral angle of 52.5° between pyridine rings.
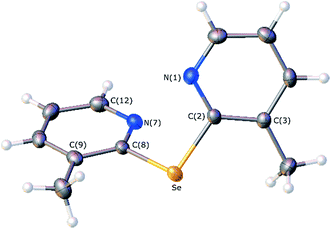 |
| | Fig. 3 Molecular structure of bis(3-methyl-2-pyridyl)selenide (2e) with thermal ellipsoids drawn at the 50% probability level. Selenium, nitrogen, carbon and hydrogen atoms are drawn in orange, light blue, grey and white, respectively. | |
Table 3 Selected bond distances (Å) and angles (°) of 2e
| Se–C(2) |
1.934(2) |
Se–C(8) |
1.933(2) |
| C(2)–Se–C(8) |
97.90(8) |
|
|
| Se–C(2)–C(3) |
119.46(15) |
Se–C(8)–C(9) |
119.43(15) |
| Se–C(2)–N(1) |
115.61(15) |
Se–C(8)–N(7) |
115.87(15) |
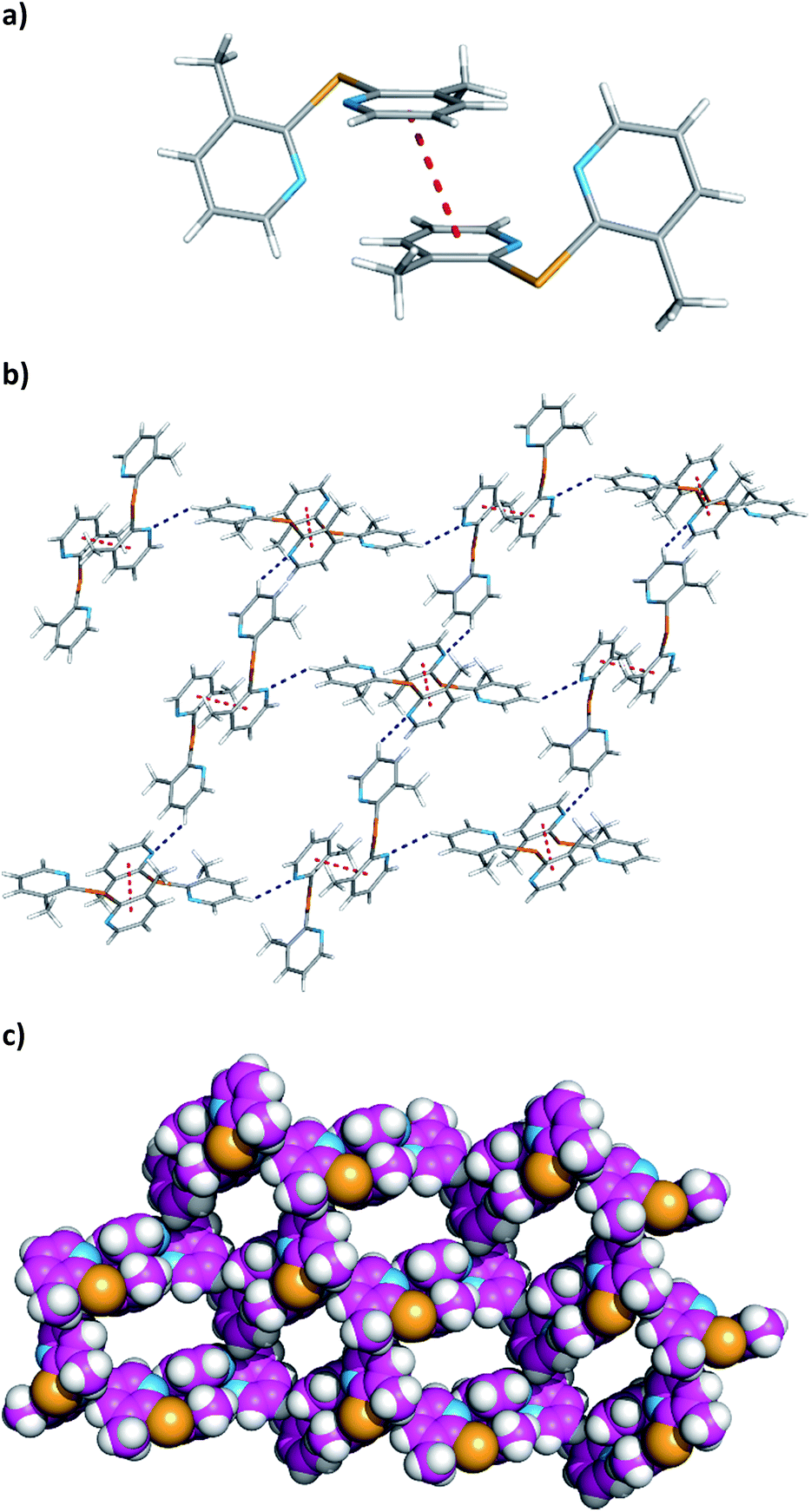
The relevant crystal packing features of 2e are depicted in Fig. 4. In the crystal lattice, two adjacent molecules are stacked over one another with a centroid–centroid distance of 3.612(2) Å and a shift distance of 1.391(3) Å. These distances are consistent with the existence of π⋯π stacking interactions resulting in the formation of a dimer as shown in Fig. 4a. The dimers are further self-assembled through C–H⋯N hydrogen bonds with C⋯N distances of 3.413(3) Å and C–H⋯N angles of 142°. Thus, the crystal structure exists as a 2-D network of π⋯π interactions and C–H⋯N hydrogen bonds. This network grows up from a ring motif composed of four dimer entities as illustrated in Fig. 4b and c.
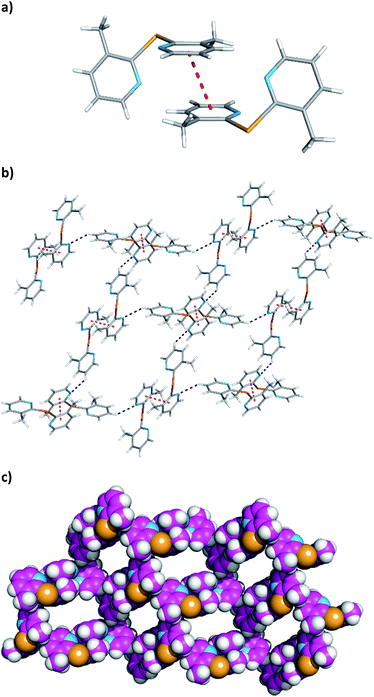 |
| | Fig. 4 Crystal packing diagram features of 2e: view (a) shows the formation of a dimer via π⋯π interactions; view (b) shows a 2-D network built up through the self-assembly of π⋯π dimer units by subsequent H⋯N hydrogen bonds (c) CPK representation of 2-D network emphasising the empty regions determined by ring motifs composed of four adjacent dimers. Colour scheme as described in Fig. 3, apart the carbon atoms in view (c), which are in light magenta. The π⋯π interactions between the pyridine rings and hydrogen bonds are drawn as red and dark blue dashed lines, respectively. | |
Anticarcinogenic activity
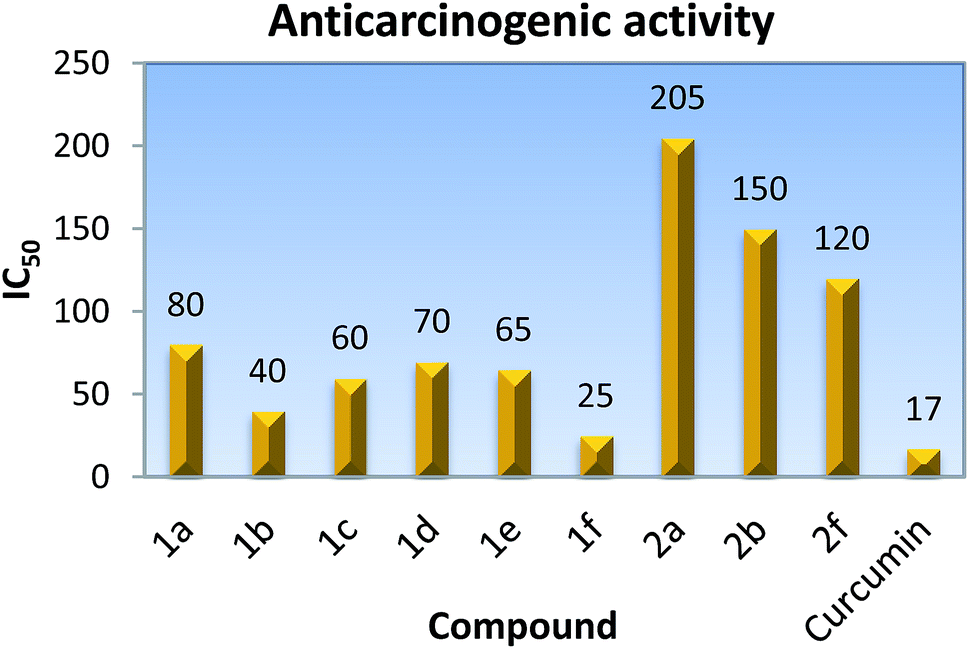
Anticarcinogenic activity of the synthesized compounds was screened at different concentrations against the Raji cancer cell line, (acute lymphoid leukemia) cells using the MTT colorimetric assay.20 The MTT assay is based on the reduction of the yellow soluble 3-(4,5-methyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) into a blue-purple formazan product, mainly by mitochondrial succinic dehydrogenase enzyme activity inside living cells. Curcumin is a known anticancer compound which has been considered as a positive control in the present study. The results were expressed as the IC50, inducing a 50% inhibition of cell growth of treated cells when compared to the growth of control cells. 1f showed the maximum activity and possessed IC50 at 25 μM. A perusal of the data in Fig. 5 reveals that the anticarcinogenic activity of the pyridylseleniums decreases with the increase in the number of methyl groups (Fig. 5). Further, it was found that the bis(2-pyridyl)diselenides are 2–5 times more active than the corresponding monoselenides, Fig. 5. The standard drug curcumin shows IC50 at about 17 μM against the Raji cancer cell line and the activity of the compound 1f is very close to it.
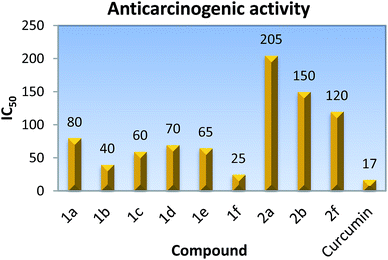 |
| | Fig. 5 IC50 values of bis(2-pyridyl)selenides and -diselenides against the Raji cancer cell line. | |
Experimental
All the experiments were carried out in a dry oxygen-free nitrogen atmosphere and the solvents were dried before use. 1H and 13C NMR spectra were recorded on a Bruker 400 MHz spectrophotometer in CDCl3. The EI mass spectra were taken by using Shimadzu GC-Mass Spectrometer [GCMS QP-2010 plus] with Rtx-1MS (30 m × 0.25 mm ID × 0.25 μm) capillary column. The elemental analysis was carried out by using Elemantar Vario MICRO analyser.
General procedure: preparation of bis(2-pyridyl)selenides
LiAlH4 (0.06 g, 1.70 mmol) was added in small installments to a vigorously stirred solution of bis(2-pyridyl)diselenide (1.30 mmol) in dry THF or DMF at −10 °C. The grayish yellow suspension was slowly warmed to room temperature and then refluxed for 12–46 h in an oil-bath. The formation of monoselenide was monitored by TLC and GC-MS. After completion, the reaction mixture was hydrolyzed with 30 mL of distilled water. The organic layer was extracted with diethyl ether (3 × 50 mL), washed with water, and then dried over anhydrous sodium sulfate. The solvent was removed and the crude product was purified by column chromatography using 60–120 mesh silica gel and hexane–ethyl acetate as an eluent (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Bis(3,5-dimethyl-2-pyridyl)selenide (2a). Diselenide:- bis(3,5-dimethyl-2-pyridyl)diselenide (0.46 g, 1.30 mmol). Yield: 0.35 g 95% (THF), 0.32 g 89% (DMF), m.p. 125–127 °C. 1H NMR: CDCl3, 400 MHz δ (ppm): 8. 14 (s, 2H), 7.18 (s, 2H), 2.41 (s, 6H), 2.24 (s, 6H). 13C NMR: CDCl3, 400 MHz δ (ppm): 149.9, 148.2, 137.9, 133.8, 131.8, 20.8, 17.7. MS (EI, 70 eV) m/z (relative intensity): 292 (18), 197 (52), 106 (22), 91 (100), 77 (24), 65 (49). Anal. calcd (%) for C14H16N2Se: C, 57.73, H, 5.53, N, 9.61. Found: C, 57.82, H, 5.51, N, 9.60.
Bis(6-methyl-2-pyridyl)selenide (2b). Diselenide:- bis(6-methyl-2-pyridyl)diselenide (0.44 g, 1.30 mmol). Yield: 0.32 g 92% (THF), 0.31 g 89% (DMF), m.p. 120–122 °C. 1H NMR: CDCl3, 400 MHz δ (ppm): 7.42 (t, 2H), 7.30–7.32 (d, J = 7.7 Hz, 2H), 6.99–7.01 (d, J = 7.4 Hz, 2H), 2.54 (s, 6H). 13C NMR: CDCl3, 400 MHz δ (ppm): 159.3, 154.4, 137.0, 124.8, 121.4, 24.3. MS (EI, 70 eV) m/z (relative intensity): 264 (100), 222 (1), 183 (48), 168 (16), 131 (6), 92 (66), 65 (100), 51 (44). Anal. calcd (%) for C12H12N2Se: C, 54.76, H, 4.59, N, 10.64. Found: C, 54.59, H, 4.55, N, 10.67.
Bis(5-methyl-2-pyridyl)selenide (2c). Diselenide:- bis(5-methyl-2-pyridyl)diselenide (0.44 g, 1.30 mmol). Yield: 0.31 g 90% (THF), 0.30 g 86% (DMF), m.p. 94–95 °C. 1H NMR: CDCl3, 400 MHz δ (ppm): 8.28–8.28 (d, J = 1.9 Hz, 2H), 7.67–7.69 (d, J = 8.1 Hz, 2H), 7.33–7.36 (d, J = 2.2, 8.1 Hz, 2H), 2.28 (s, 3H). 13C NMR: CDCl3, 400 MHz δ (ppm): 150.9, 149.8, 138.2, 130.9, 123.4, 17.8. MS (EI, 70 eV) m/z (relative intensity): 263 (76), 247 (1), 183 (6), 168 (3), 93 (39), 65 (100), 53 (5). Anal. calcd (%) for C12H12N2Se: C, 54.76, H, 4.59, N, 10.64. Found: C, 55.64, H, 4.53, N, 10.61.
Bis(4-methyl-2-pyridyl)selenide (2d). Diselenide:- bis(4-methyl-2-pyridyl)diselenide (0.44 g, 1.30 mmol). Yield: 0.31 g 91% (THF), 0.29 g 84% (DMF), m.p. 130–132 °C. 1H NMR: CDCl3, 400 MHz δ (ppm): 8.29–8.31 (d, J = 5.0 Hz, 2H), 7.61 (s, 2H), 6.88–6.89 (d, J = 5.1 Hz, 2H), 2.28 (s, 6H). 13C NMR: CDCl3, 400 MHz δ (ppm): 154.2, 149.1, 123.9, 122.4, 96.1, 21.1. MS (EI, 70 eV) m/z (relative intensity): 264 (39), 243 (89), 249 (73), 183 (13), 168 (14), 132 (57), 92 (33), 65 (100), 51 (64). Anal. calcd (%) for C12H12N2Se: C, 54.76, H, 4.59, N, 10.64. Found: C, 54.73, H, 4.53, N, 10.69.
Bis(3-methyl-2-pyridyl)selenide (2e). Diselenide:- bis(3-methyl-2-pyridyl)diselenide (0.44 g, 1.30 mmol). Yield: 0.30 g 90% (THF), 0.29 g 83% (DMF), m.p. 177–182 °C. 1H NMR: (CDCl3, 400 MHz): δ (ppm): 8.47–8.49 (dd, J = 1.9, 4.7 Hz, 2H), 7.38–7.41 (dd, J = 4.7, 7.5 Hz, 2H), 6.87 (t, 2H), 2.19 (s, 6H). 13C NMR: (CDCl3, 400 MHz): δ (ppm): 140.3, 138.4, 132.7, 125.0, 120.3, 25.7. MS (EI, 70 eV) m/z (relative intensity): 266 (8), 264 (41), 249 (65), 183 (100), 172 (6), 168 (40), 51 (9). Anal. calcd (%) for C12H12N2Se: C, 54.76, H, 4.59, N, 10.64. Found: C, 54.88, H, 4.51, N, 10.60.
Bis(2-pyridyl)selenide (2f)4c. Diselenide:- bis(2-pyridyl)diselenide (0.40 g, 1.30 mmol). Yield: 0.29 g 96% (THF), 0.27 g 88% (DMF), m.p. 93–94 °C. 1H NMR: CDCl3, 400 MHz δ (ppm): 8.53–8.54 (ddd, J = 1.5, 2.6, 4.8 Hz, 2H), 7.54–7.59 (m, 2H), 7.14–7.19 (m, 2H). 13C NMR: CDCl3, 400 MHz δ (ppm): 155.0, 150.4, 137.0, 128.1, 121.9. MS (EI, 70 eV) m/z (relative intensity): 235 (42), 155 (7), 78 (98), 51 (100). Anal. calcd (%) for C10H8N2Se: C, 51.07, H, 3.42, N, 8.01. Found: C, 51.12, H, 3.48, N, 7.97.
Synthesis of selenides from elemental selenium and LiAlH4
Selenium (1 g, 12.66 mmol) powder and dry DMF were taken in a three necked 100 mL RBF. The temperature of the reaction mixture was lowered to −10 °C and LiAlH4 (0.48 g, 12.66 mmol) was added in small instalments under dry oxygen free nitrogen atmosphere. The grayish suspension was formed and slowly brought to the room temperature. The reaction mixture was stirred at room temperature for 30 min and again re-cooled to 0 °C and then 2-bromopyridine (12.66 mmol) was added to it in a drop-wise manner. The reaction mixture was slowly brought to the room temperature and then, refluxed for 3 h at 140 °C. The process of reaction was monitored by TLC. After the completion of reaction, the solvent was removed under vacuum with the help of a rota-evaporated. The solid residue was dissolved in chloroform (50 mL) and the organic layer was washed with water. The organic layer was then dried over anhydrous sodium sulfate. The solvent was removed and the crude product was purified by column chromatography using 60–120 mesh silica gel and hexane–ethyl acetate as an eluent. Following the same procedure, other bis(2-pyridyl)selenides and bis(2-pyridyl)diselenides were also prepared.
Bis(6-methyl-2-pyridyl)diselenide (1b)5a. M.p. 61–62 °C. 1H NMR: (CDCl3, 400 MHz): δ (ppm): 7.53–7.55 (d, J = 8.3 Hz, 2H), 7.39–7.41 (d, J = 8.3 Hz, 1H), 2.55 (s, 3H). 13C NMR: (CDCl3, 400 MHz): δ (ppm): 150.8, 149.8, 138.2, 130.9, 123.4, 17.8. MS (ESI): 344 (80Se). Anal. calcd (%) for C12H12N2Se2: C, 42.12, H, 3.53, N, 8.18. Found: C, 41.85, H, 3.59, N, 8.08.
Bis(5-methyl-2-pyridyl)diselenide (1c)5a. M.p. 65–66 °C. 1H NMR: CDCl3, 400 MHz δ (ppm): 8.28 (s, 2H), 7.68–7.70 (d, J = 8.1 Hz, 2H), 7.34–7.36 (dd, J = 1.9, 8.1 Hz, 2H), 2.29 (s, 6H). 13C NMR: (CDCl3, 400 MHz): δ (ppm): 150.8, 149.8, 138.2, 130.9, 123.4, 17.8. MS (ESI): 344 (80Se). Anal. calcd (%) for C12H12N2Se2: C, 42.12, H, 3.53, N, 8.18. Found: C, 41.87, H, 3.56, N, 8.10.
Bis(4-methyl-2-pyridyl)diselenide (1d)5a. M.p. 96–98 °C. 1H NMR: (CDCl3, 400 MHz): δ (ppm): 8.31–8.29 (d, J = 4.9 Hz, 2H), 7.65 (s, 2H), 6.90–6.91 (d, J = 4.1 Hz, 2H), 2.29 (s, 6H). 13C NMR: (CDCl3, 400 MHz): δ (ppm): 154.1, 149.1, 148.9, 123.9, 122.4, 21.0. MS (EI, 70 eV) m/z (relative intensity): 344 (10), 263 (5), 183 (59), 92 (99), 80 (6), 65 (100), 51 (7). Anal. calcd (%) for C12H12N2Se2: C, 42.12, H, 3.53, N, 8.18. Found: C, 42.21, H, 3.40, N, 8.03.
Bis(3-methyl-2-pyridyl)diselenide (1e)5a. M.p. 142–144 °C. 1H NMR: (CDCl3, 400 MHz): δ (ppm): 8.28–8.30 (d, J = 5.9 Hz, 2H), 7.26–7.34 (d, J = 7.4 Hz, 2H), 7.00–7.03 (t, J = 7.4 Hz, 2H), 2.46 (s, 6H). 13C NMR: (CDCl3, 400 MHz): δ (ppm): 153.3, 147.9, 136.8, 133.7, 121.8, 20.7. MS (EI, 70 eV) m/z (relative intensity): 264 (42), 249 (65), 183 (100), 168 (40), 131 (8), 92 (29), 65 (4), 51 (9). Anal. calcd (%) for C12H12N2Se2: C, 54.76, H, 4.59, N, 10.64. Found: C, 54.86, H, 4.51, N, 10.76.
Bis(2-pyridyl)diselenide (1f)21. M.p. 48–50 °C. 1H NMR: (CDCl3, 400 MHz): δ (ppm): 8.44–8.46 (dd, J = 1.7, 7.4 Hz, 2H), 7.78–7.80 (d, J = 7.1 Hz), 7.51–7.56 (m, 2H), 7.06–7.09 (td, J = 1.0, 7.3 Hz, 2H). 13C NMR: (CDCl3, 400 MHz): δ (ppm): 154.4, 149.6, 137.5, 123.5, 121.2. MS (EI, 70 eV) m/z (relative intensity): 235 (42), 155 (7), 78 (98), 51 (100). Anal. calcd (%) for C10H8N2Se2: C, 51.07, H, 3.42, N, 8.01. Found: C, 51.12, H, 3.48, N, 7.97.
Crystallography
The single crystal X-ray data of 2e was collected on a Bruker SMART Apex II CCD-based diffractometer at 180(2) K, using graphite monochromatized Mo-Kα radiation (λ = 0.71073 Å). Data reduction was carried out using the SAINT-NT software package from Bruker AXS.22 The raw intensities were corrected for absorption effects through the multi-scan method with SADABS.23 The structure was solved by a combination of direct methods and subsequent difference Fourier syntheses and refined by full matrix least squares on F2 using the SHELX-2013 suite.24 The hydrogen atoms were inserted at geometrical positions. Anisotropic thermal parameters were used for all non-hydrogen atoms, while the C–H hydrogen atoms were refined with isotropic parameters equivalent 1.2 times of the atoms to which they are attached. Molecular and crystal packing diagrams were drawn with Olex2 (ref. 25) and PyMOL, respectively.26 The crystal data and refinement details of 2e are summarized in the Table 4.
Table 4 Crystal data and relevant refinement details of 2e
| Empirical formula |
C12H12N2Se |
| Mw |
263.20 |
| Crystal system |
Monoclinic |
| Space group |
P21/n |
| a/Å |
7.5073(3) |
| b/Å |
19.2645(59) |
| c/Å |
7.5241(4) |
| β/° |
92.684(2) |
| V/Å3 |
1086.97(9) |
| Z |
4 |
| Dc/g cm−3 |
1.608 |
| μ/mm−1 |
3.420 |
| F(000) |
528 |
| 2θ range/° |
4.23–58.28 |
| Index ranges |
−10 ≤ h ≤ 9, −21 ≤ k ≤ 26, −10 ≤ l ≤ 10 |
| Reflections collected |
17964 |
| Unique reflections, [Rint] |
2930 [0.0501] |
| Goodness-of-fit on F2 |
1.020 |
| Final R indices |
|
| R1, wR2, [I > 2σI] |
0.0306, 0.0583 [2287] |
| R1, wR2 (all data) |
0.0481, 0.0640 |
| Largest diff. peak/hole/eÅ−3 |
0.51/−0.44 |
Anticarcinogenic activity
Anticarcinogenic activity of the synthesized compounds was evaluated against the Raji cancer cell line using the MTT colorimetric assay.20 The stock solutions of the synthesized compounds were prepared by dissolving in the minimum volume of dimethyl sulfoxide (DMSO), which was further diluted to reach the appropriate volume. Cells (1 × 104 cells per well) were plated in 96-multiwell plates. After incubation for 48 h, MTT dissolved in phosphate buffer saline (PBS) was added to each well at a final concentration of 5 mg mL−1 and then incubated at 37 °C and 5% CO2 for 2 h. The Raji (acute lymphoid leukemia) cells were maintained in a humidified atmosphere of 5% CO2, 95% air at 37 °C. Curcumin was used as a positive control and the wells containing DMSO (1%) and cell suspension was regarded as the negative control. The blank wells were consisted of 200 μL of the RPMI medium. The microplates were further incubated for 48 h. To evaluate cell survival, each well was then incubated with 20 μL of MTT solution (5 mg mL−1 in PBS) and incubated for 4 h at 37 °C. The media in each well was replaced with 200 μL DMSO and pipetted up and down to dissolve the formazan crystals which were formed by the cellular reduction of MTT. The absorbance of each well was measured at 570 nm using an ELISA reader. Each experiment was repeated three times. The results were expressed as the IC50, which induces a 50% inhibition of cell growth of treated cells when compared to the growth of control cells.
Computational details
The entire quantum calculations were performed by using Gaussian 03 (ref. 27) program on a Pentium personal computer. Complete geometry optimizations were performed by density functional theoretical (DFT) computations by using the closed-shell Becke–Lee–Yang–Parr hybrid exchange–correlation three-parameter functional (B3LYP) in combination with 6-31 + G(d,p) basis set.28
Conclusions
The discussed protocol affords analytically pure symmetrical monoselenide without any contamination of the respective diselenide. The reaction involves the formation of a selenated aluminato complex that undergoes pyridine-shift reaction leading to the generation of LiAlSeH2 and symmetrical bis(2-pyridyl)selenide. The proposed mechanism has been validated by experimental observations, 1H NMR analysis and theoretical analysis. The theoretical analysis showed a showed strong tendency of the reaction between LiAlH4 and bis(2-pyridyl)diselenides leading to the formation of complex (I). The synthesized compounds were also evaluated as anticarcinogenic agents and compound 1f showed anticarcinogenic activity comparable to the standard drug curcumin.
Acknowledgements
We are thankful to Mr Avtar Singh, CIL, Punjab University, Chandigarh, for NMR spectra.
Notes and references
- D. M. Freudendahl, S. A. Shahzad and T. Wirth, Eur. J. Org. Chem., 2009, 42, 1649 CrossRef PubMed.
-
(a) J. Wang, P. Liu, C. C. Seaton and K. M. Ryan, J. Am. Chem. Soc., 2014, 136, 7954 CrossRef CAS PubMed;
(b) J. Gu, Z.-Q. Zhao, Y. Ding, H.-L. Chen, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 8363 CrossRef CAS PubMed.
-
(a) A. Molter, G. N. KaluCerović, H. Kommera, R. Paschke, T. Langer, R. Pöttgen and F. Mohr, J. Organomet. Chem., 2012, 701, 80 CrossRef CAS PubMed;
(b) Y. A. Lin, O. Boutureira, L. Lercher, B. Bhushan, R. S. Paton and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 12156 CrossRef CAS PubMed;
(c) M. R. Nicole, N. M. R. McNeil, M. C. Matz and T. G. Back, J. Org. Chem., 2013, 78, 10369 CrossRef PubMed;
(d) F. Martínez-Ramos, H. Salgado-Zamora, M. E. Campos-Aldrete, E. Melendez-Camargo, Y. Márquez-Flores and M. Soriano-García, Eur. J. Med. Chem., 2008, 43, 1432 CrossRef PubMed;
(e) P. Arsenyan, K. Rubina, I. Shestakova and I. Domracheva, Eur. J. Med. Chem., 2007, 42, 635 CrossRef CAS PubMed;
(f) J. S. Dhau, A. Singh, A. Singh and B. S. Sooch, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 687 CrossRef CAS PubMed.
-
(a) K. Shimada, S. Oikawa, H. Nakamura, K. Moro-oka, M. Kikuchi, A. Maruyama, T. Suzuki, H. Kogawa, Y. Inoue, Y. Gong, S. Aoyagi and Y. Takikawa, Bull. Chem. Soc. Jpn., 2005, 78, 899 CrossRef CAS;
(b) K. K. Bhasin, V. K. Jain, H. Kumar, S. Sharma, S. K. Mehta and J. Singh, Synth. Commun., 2003, 33, 977 CrossRef CAS PubMed;
(c) A. S. Hodage, P. C. Parashiva, P. P. Phadnis, A. Wadawale, K. I. Priyadarsini and V. K. Jain, J. Organomet. Chem., 2012, 720, 19 CrossRef CAS PubMed;
(d) F. M. Libero, M. C. D. Xavier, F. N. Victoria, P. S. Nascente, L. Savegnago, G. Perin and G. Alves, Tetrahedron Lett., 2012, 53, 3091 CrossRef CAS PubMed;
(e) A. L. Braga, W. A. S. Filho, R. S. Schwab, O. E. D. Rodrigues, L. Dornelles, H. C. Braga and D. S. Lüdtke, Tetrahedron Lett., 2009, 50, 3005 CrossRef CAS PubMed;
(f) R. K. Sharma, G. Kedarnath, A. Wadawale, C. A. Betty, B. Vishwanadh and V. K. Jain, Dalton Trans., 2012, 41, 12129 RSC;
(g) R. K. Sharma, G. Kedarnath, V. K. Jain, A. Wadawale, C. G. S. Pillai, M. Nalliath and B. Vishwanadh, Dalton Trans., 2011, 40, 9194 RSC.
-
(a) K. K. Bhasin and J. Singh, J. Organomet. Chem., 2002, 658, 71 CrossRef CAS;
(b) E. N. Deryagina, N. V. Russavskaya, L. K. Papernaya, E. P. Levanova, E. N. Sukhomazova and N. A. Korchevin, Russ. Chem. Bull., 2005, 54, 2473 CrossRef CAS.
-
(a) J. S. Dhau, A. Singh, Y. Kasetti and P. V. Bharatam, Eur. J. Org. Chem., 2012, 1746 CrossRef CAS PubMed;
(b) J. S. Dhau, A. Singh, Y. Kasetti, S. Bhatia, P. V. Bharatam, P. Brandão, V. Félix and K. N. Singh, Tetrahedron, 2013, 69, 10284 CrossRef CAS PubMed;
(c) J. S. Dhau, A. Singh and R. Dhir, J. Organomet. Chem., 2011, 696, 2008 CrossRef CAS PubMed.
- S. Kumar, H. Johansson, L. Engman, L. Valgimigli, R. Amorati, M. G. Fumo and G. F. Pedulli, J. Org. Chem., 2007, 72, 2583 CrossRef CAS PubMed.
- J.-M. Becht and C. L. Drian, J. Org. Chem., 2011, 76, 6327 CrossRef CAS PubMed.
- N. Mukherjee, T. Chatterjee, C. Brindaban and B. C. Ranu, J. Org. Chem., 2013, 78, 11110 CrossRef CAS PubMed.
- V. P. Reddy, A. V. Kumar and K. R. Rao, J. Org. Chem., 2010, 75, 8720 CrossRef CAS PubMed.
- H. Ishihara, M. Koketsu, Y. Fukuta and F. Nada, J. Am. Chem. Soc., 2001, 123, 8408 CrossRef CAS.
-
(a) J. S. Dhau, A. Singh, A. Singh, B. S. Sooch, P. Brandão and V. Félix, Inorg. Chim. Acta, 2012, 392, 335 CrossRef CAS PubMed;
(b) J. S. Dhau, R. Dhir and A. Singh, J. Organomet. Chem., 2011, 696, 2406 CrossRef CAS PubMed.
- G. Zhao, H. Lin, S. Zhu, H. Sun and Y. Chen, Anti-Cancer Drug Des., 1998, 13, 767 Search PubMed.
- K. K. Bhasin, Rishu, S. Singh, H. Kumar and S. K. Mehta, J. Organomet. Chem., 2010, 695, 648 CrossRef CAS PubMed.
-
(a) J. S. Dhau, R. Dhir, A. Singh, A. Singh, P. Brandão and V. Félix, Tetrahedron, 2014, 70, 4876 CrossRef CAS PubMed;
(b) A. Singh, J. S. Dhau, N. Sharma and A. Singh, Inorg. Chim. Acta, 2015, 432, 109 CrossRef CAS PubMed;
(c) J. S. Dhau, A. Singh, A. Singh, B. S. Sooch, P. Brandão and V. Félix, J. Organomet. Chem., 2014, 766, 57 CrossRef CAS PubMed.
- J. A. Gladysz, J. L. Hornby and J. E. Garbe, J. Org. Chem., 1978, 43, 1204 CrossRef CAS.
- N. Sklar and B. Post, Inorg. Chem., 1967, 6, 699 CrossRef.
-
(a) H. G. Mautner, S.-H. Chu and C. M. Lee, J. Org. Chem., 1962, 27, 3671 CrossRef CAS;
(b) A. Toshimitsu, S. Owada, S. Uemura and M. Okano, Tetrahedron Lett., 1980, 21, 5037 CrossRef CAS;
(c) K. Smith, I. Matthews, N. M. Hulme and G. E. Martin, J. Chem. Soc., Perkin Trans. 1, 1986, 2075 RSC;
(d) Y. Cheng, J. Emge and J. G. Brennan, Inorg. Chem., 1994, 33, 3711 CrossRef CAS.
- S. J. Dunne, E. I. von Nagy-Felsobuki and M. F. Mackay, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1454 CrossRef.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley and P. Cronise, J. Natl. Cancer Inst., 1991, 83, 757 CrossRef CAS PubMed.
- C. O. Kienitz, C. Thöne and P. G. Jones, Inorg. Chem., 1996, 35, 3990 CrossRef CAS PubMed.
- Bruker SAINT-plus, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- G. M. Sheldrick, SADABS, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- PyMOL Molecular Graphics System, Version 1.2r2, DeLano Scientific LLC, 2009 Search PubMed.
- M. J. Frisch, et al., GAUSSIAN 03 Program, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.