
Double helicity induction in chiral bis(triphenylacetamides)†
Natalia Prusinowskaa,
Wioletta Bendzińska-Berusa,
Joanna Szymkowiakab,
Beata Warżajtisa,
Jadwiga Gajewyab,
Maciej Jeleckiab,
Urszula Rychlewska*a and
Marcin Kwit*ac
aDepartment of Chemistry, Adam Mickiewicz University, Umultowska 89B, 61 614 Poznań, Poland. E-mail: urszular@amu.edu.pl; marcin.kwit@amu.edu.pl
bWielkopolska Center for Advanced Technologies (WCAT), Umultowska 89C, 61 614 Poznań, Poland
cInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 25th September 2015
Abstract
A combination of experimental methods (ECD, X-ray diffraction) and theoretical calculations allowed the description of chirality transfer in bis(triphenylacetamides). For the first time it has been shown that effective helicity induction in a trityl chromophore is not only due to the presence of stereogenic center(s) but is also caused by other chirality inductors such as an axis of chirality. For all experimental and computed ECD spectra a uniformly identical sequence of signs of the Cotton effects has been found: negative at around 215 nm and positive below 200 nm. This result can only be explained by the dependence of the ECD spectra on the R absolute configuration of the proximal stereogenic element. The chirality transfer is achieved through a series of weak intramolecular interactions. Helicities of the trityl chromophores and their propeller shapes are influenced by the structure of the chiral inductor and the steric hindrance at amide nitrogen atoms. The stereogenic centers in the diamide unit act independently as chirality inductors and the direct trityl⋯trityl interactions are only rarely observed. Characteristic for the investigated crystals is the complete hindrance of the amide NH groups and their inability to be involved in any intermolecular interactions as well as their highly restricted ability for the formation of intramolecular hydrogen bonds. This results from the presence of Tr groups, which act as supramolecular NH protecting groups, especially when attached to the rigid carbocycles.
Introduction
The trityl (triphenylmethyl, Tr) group, a widely used protecting group in organic synthesis,1 in recent years has received attention as a component of molecular devices,2 catalysts3 and chirality reporters.4 The latter application has been found to be closely related to the structural and dynamic properties of triphenylmethane derivatives. The Tr group connected to the achiral spherical substituent formed a dynamically racemic propeller of formal C3 symmetry, which did not generate an electronic circular dichroism (ECD) signal.5 Introduction of any chiral substituent in close proximity of the trityl group shifted the conformational equilibrium into energetically preferred diastereomer(s). The transmission of chirality from the stereogenic center to the trityl group through a molecular “bevel gear” mechanism was demonstrated for the first time by Gawronski et al.4 The trityl group can act not only as a molecular rotator but it demonstrates an ability to rotation of each phenyl group (slippage). For this reason, the macroscopic equivalent of Tr group could rather be a ‘helicopter’ airscrew than a boat power screw.DFT calculations showed that the Tr group in chiral molecules did not form a regular C3-symmetry propeller, but adopted the lowest (C1) symmetry, with a low-energy barrier of a two-ring flip through a Cs-symmetry transition state.4
Similarly to the previously reported O-tritylalcohols and N-tritylamines, the transmission of chirality from the stereogenic centers to the reporter units was observed also for ditrityl substituted molecules being derivatives of cyclic chiral 1,2-diols and 1,2-diamines.6 Due to higher conformational flexibility of ditrityl derivatives, correlation between structure and chiroptical properties was not straightforward. For trans-1,2-disubstituted derivatives of cyclohexane and their five-membered ring congeners, conformational equilibria involved structures in which X–Tr substituents were either equatorial or axial, depending on substituent X. When X = O, a diaxial conformer predominated, whereas in the case of X = NH a diequatorial conformer was more stable due to the presence of intramolecular N–H⋯N hydrogen bond.
Recently, we have shown that elongation of linker between the inductor (the stereogenic center) and the reporter (the trityl chromophore) via an amide unit had profound influence on the chirality transmission and the conformational equilibria.7 The triphenylacetic acid chromophore displayed very high sensitivity to even subtle changes in the size of the substituent at the stereogenic center. The comparable sensitivity in chirogenesis was reported so far for systems based on bis(zincporphyrin) and N-(1,8-naphthaloyl)-2-aminobenzoyl (NAB) derivatives.8 The optical activity model proposed for the aliphatic triphenylacetamides correlated absolute configuration at the stereogenic center with a helicity of the reporter unit. For secondary (S)-amides the conformation of the trityl group was found by calculations as MMM in the gas phase. In tertiary (S)-amides the conformational equilibria were shifted into OPP or OMM diastereoisomers.7 The process of chirality transmission in the triphenylacetamides was achieved solely through weak and cooperative intramolecular interactions between C–H/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(amide) local dipoles. However, X-ray studies revealed that some of the chiral triphenylacetamides crystallized with two symmetry independent molecules that differed only in the helicity of Tr moieties, giving rise to conformational enantiomerism and configurational/conformational diastereoisomerism.
O(amide) local dipoles. However, X-ray studies revealed that some of the chiral triphenylacetamides crystallized with two symmetry independent molecules that differed only in the helicity of Tr moieties, giving rise to conformational enantiomerism and configurational/conformational diastereoisomerism.
Since the concept of the chirality transmission in the triphenylacetamides is still relatively unexplored we decided to expand our studies to a novel class of triphenylacetamides, namely bis(triphenylacetamides), where Tr chromophores are attached to either cyclic or non-cyclic skeletons bearing vicinal amine substituents. We have expected the following events to occur (i) different chiroptical response, as compared to monotritylacetamides due to the presence of two bulky substituents in proximity; (ii) helicity and chiroptical response of a given reporter unit resulting solely from inductor–receptor interactions (“independent” model); (iii) helicity and chiroptical response of a given reporter unit resulting mainly from intramolecular trityl⋯trityl interactions; (iv) the object to its mirror-image relationship between trityl groups attached to the same skeleton (based on the analogy with the pseudocentrosymmetric crystal structures of monoamides); (v) effective induction of helicity within the chromophore from other than point stereogenic elements. We have anticipated that the model of optical activity proposed for mono triphenylacetamides may no longer be valid for the disubstituted compounds.
Materials and methods
Bis(triphenylacetamides) 1–8 (Fig. 1) were obtained in moderate to high yields by reactions of respective primary or secondary diamines with triphenylacetoyl chloride (detailed experimental procedures and full spectroscopic characterization for all new compounds were deposited in Experimental Section in ESI†). Crystals suitable for X-ray diffraction were obtained by slow evaporation of diethyl ether or dichloromethane from ether-n-hexane or dichloromethane-n-hexane solution of respective amide. To show a possible environmental effect on the ECD spectra of amides 1–8 we used solvents of different polarity: cyclohexane and acetonitrile.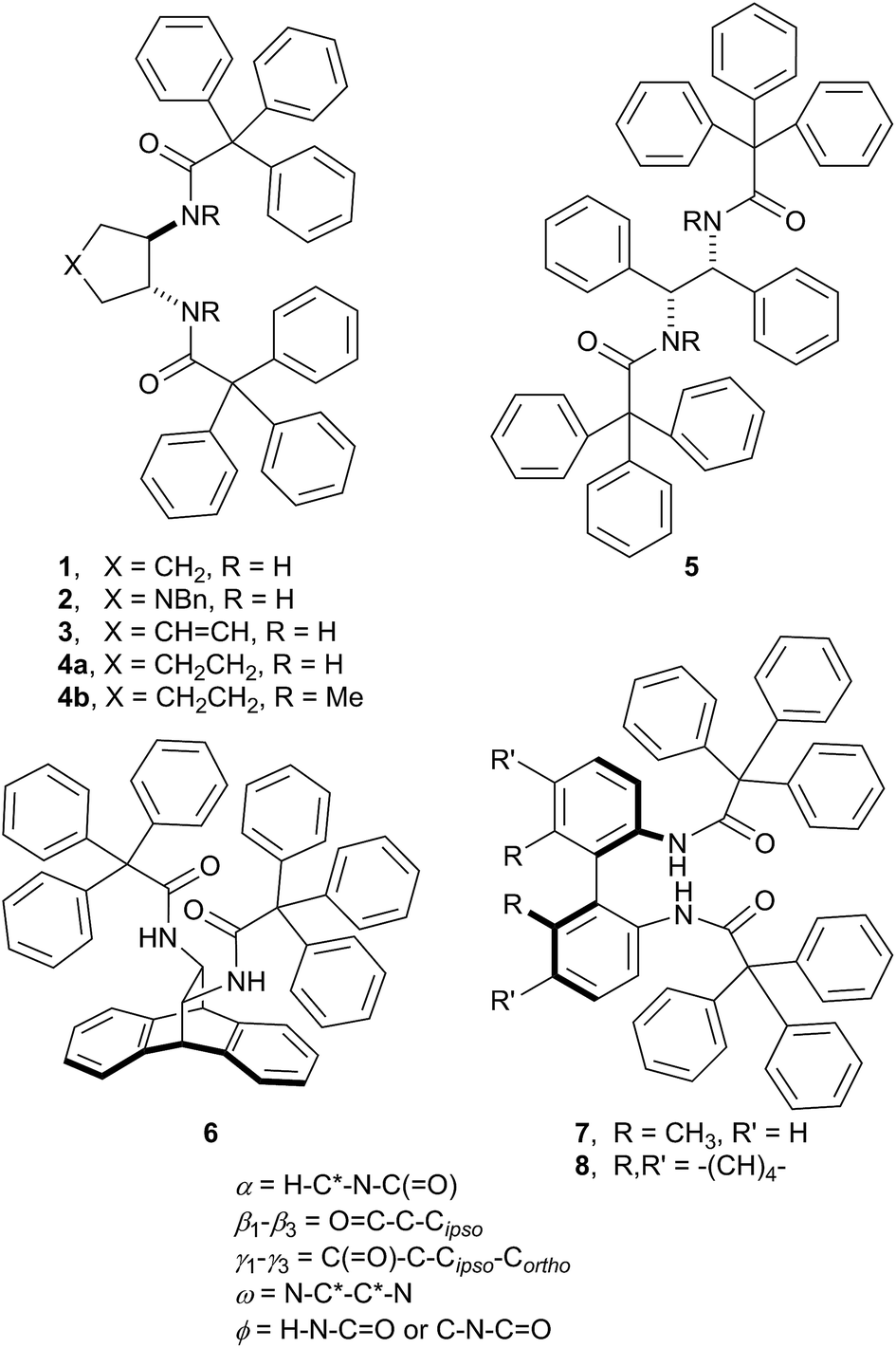 | ||
| Fig. 1 Structures of bis(triphenylacetamides) 1–8 and definitions of the structural parameters that characterize conformation of the molecule. | ||
Computational studies of the chirality transmission mechanisms in the selected bis(triphenylacetamides) followed the previously proposed protocol.7 This included systematical conformational searches at molecular mechanics level,9 where all rotatable torsion angles were changed in 30° steps; pre-optimization of all conformers at the B3LYP/6-31G(d)10 level followed by higher accuracy calculations. Since it is believed that the structure of compounds having CPh3 groups in close proximity is affected by non-covalent dispersive interactions,11 we used pure B97 (ref. 12) and B3LYP,13 M06-2X14 hybrid functionals, including (or not) the empirical long-range corrections,15 all in conjunction with the enhanced 6-311G(2d,2p) basis set.10 This allowed us to reveal the role of dispersive interactions as factors that may influence the overall shape of the chiral ditrityl-substituted molecules. Generally, addition of empirical correction reduced the number of low-energy conformers. This effect was more visible for “old” B3LYP than for the newer M06-2X hybrid functional. Originally, the M06-2X functional was constructed, among others, for better reproduction of aromatic–aromatic stacking interactions,14 thus, the empirical correction for these interactions had only negligible effect. The structures calculated with the use of D3 empirical corrections are more “compact” – the trityl groups were closer to each other than in the structures calculated without correction. The change in geometry was associated with the change of abundance of a given conformer. For example, when the B3LYP-GD3BJ method was used, the population of the conformer no 7 of amide 1 amounted to 54% and was reduced to 3% only, when the same functional without dispersion correction was employed.
For ECD calculations only the real minimum energy conformers that differed from the most stable one by less than 2 kcal mol−1 were taken into consideration, following a generally accepted protocol. Rotatory strengths for all structures optimized at the DFT/6-311G(2d,2p) level were calculated employing B3LYP,13 CAM-B3LYP,16 B2LYP,17 LC-wPBE18 and M06-2X14 hybrid functionals, all in conjunction with 6-311G(2d,2p) basis set.10 The rotatory strengths were calculated using both length and velocity representations. Due to the negligible differences between the length and velocity calculated values, only the velocity representations were used in the present study. The ECD spectra were simulated by overlapping Gaussian functions for each transition, according to the procedure previously described by Harada and Stephens.19 The choice of the best method of geometry/energy determination has been done by comparing the Boltzmann-averaged calculated ECD with the experimental data.20 In the case of bis(triphenylacetamides) 1, 4a, 4b, 6 and 7, the TD-M06-2X/6-311G(2d,2p)//B3LYP/6-311G(2d,2p) method was found as the best for geometry-ECD calculations, whereas in the case of 5 the TD-M06-2X/6-311G(2d,2p)//M06-2X/6-311G(2d,2p) combination gave the results slightly better than others. The relative energies (unit kcal mol−1) and structures discussed here refer to Gibbs free energies (ΔΔG°) and geometries computed at the B3LYP/6-311G(2d,2p) or M06-2X/6-311G(2d,2p) levels.
All computational results and short comments on the performance of the methods were deposited in the ESI (Tables A1–B6, Fig. A1–H6†).
X-ray diffraction studies have been performed at normal conditions for compounds 4b and 5 and at 130 K for compounds 1, 4a and 6. Reflection intensities for crystals 1, 4a, 4b and 5 were measured on a SuperNova diffractometer equipped with a Cu microfocus source. Diffraction data for 6 was measured with a Xcalibur diffractometer and monochromated Mo Kα radiation. Data reduction and analysis for all structures were carried out with CrysAlisPro.21 The structures have been determined and refined with SHELX programs.22 Interpretation of the results has been performed using Mercury program.23 Crystal data are provided in the ESI (Table S1).† Fig. S1 of ESI† displays molecular conformations present in crystals.
Molecular structure in silico and in crystal
The conformation of the bis(triphenylacetamides) was described conveniently by a set of torsion angles: α, β, γ and ω (see Fig. 1 for the definition). The angles α (H–C*–N–C(O)) and β1–β3 (OC–C–Cipso) characterized the spatial arrangement of atoms around the amide groups (see Fig. 1).
The conformation of the trityl group was described by a set of three twist angles γ1–γ3 (C(O)–C–Cipso–Cortho) lying in the range of −90 to 90°, where γ1 corresponds to the lowest and γ3 to the highest absolute value of γ.7 Qualitatively, the conformation of each phenyl ring in a given trityl group was defined by its helicity which can be either M (−90° < γ < 0°), P (0° < γ < 90°) or O (for γ angles deviating from planarity by ±5°). Torsion angle ω (N–C*–C*–N) defined the mutual orientation of nitrogen atoms.
The relative energies calculated at the DFT/6-311G(2d,2p) level and percentage populations of individual conformers for representative amides 1, 4–7 were collected in Table 1. The experimental and computational data that characterize the lowest energy structures of bis(triphenylamides) 1, 4–7 were collected in Table 2 (the full version of Table 2 was deposited as ESI†). Fig. 2 shows exemplary structures of the calculated lowest-energy conformers.
| Amidea | Energy | ΔE | Pop. | ΔΔG° | Pop. |
|---|---|---|---|---|---|
| a Conformers are numbered according to their appearance during conformational search.b Calculated at the M06-2X/6-311G(2d,2p) level. | |||||
| 1 (conf. 1) | −1999.337412 | 0.00 | 40 | 0.00 | 56 |
| 1 (conf. 3) | −1999.335676 | 1.09 | 6 | 3.08 | — |
| 1 (conf. 4) | −1999.337000 | 0.27 | 26 | 0.67 | 18 |
| 1 (conf. 5) | −1999.336238 | 0.74 | 12 | 0.69 | 18 |
| 1 (conf. 6) | −1999.335284 | 1.34 | 4 | 1.50 | 5 |
| 1 (conf. 7) | −1999.334336 | 1.93 | 2 | 1.71 | 3 |
| 1 (conf. 8) | −1999.336085 | 0.83 | 10 | 2.1 | — |
| 4a (conf. 1) | −2038.670358 | 0.54 | 29 | 1.19 | 12 |
| 4a (conf. 3) | −2038.671223 | 0.00 | 71 | 0.00 | 88 |
| 4b (conf. 1) | −2117.26568 | 0.03 | 44 | 0.08 | 41 |
| 4b (conf. 5) | −2117.26427 | 0.92 | 10 | 1.05 | 8 |
| 4b (conf. 7) | −2117.26226 | 2.18 | — | 1.69 | 3 |
| 4b (conf. 8) | −2117.26573 | 0.00 | 46 | 0.00 | 48 |
| 5 (conf. 1)b | −2343.865626 | 0.96 | 10 | 1.41 | 6 |
| 5 (conf. 5)b | −2343.866888 | 0.17 | 38 | 0.52 | 25 |
| 5 (conf. 60)b | −2343.867159 | 0.00 | 52 | 0.00 | 60 |
| 5 (conf. 61)b | −2343.859347 | 4.90 | — | 1.85 | 3 |
| 5 (conf. 69)b | −2343.863000 | 2.67 | — | 1.37 | 6 |
| 6 (conf. 1) | −2421.021116 | 0.04 | 17 | 1.19 | 4 |
| 6 (conf. 31) | −2421.020526 | 0.41 | 9 | 1.35 | 3 |
| 6 (conf. 32) | −2421.020666 | 0.32 | 11 | 1.55 | 2 |
| 6 (conf. 37) | −2421.021176 | 0.00 | 19 | 1.87 | 1 |
| 6 (conf. 62) | −2421.018833 | 1.47 | 2 | 2.69 | — |
| 6 (conf. 68) | −2421.020267 | 0.57 | 7 | 0.03 | 30 |
| 6 (conf. 70) | −2421.021175 | 0.00 | 19 | 0.75 | 9 |
| 6 (conf. 73) | −2421.020650 | 0.33 | 11 | 0.81 | 8 |
| 6 (conf. 81) | −2421.019186 | 1.25 | 2 | 0.77 | 9 |
| 6 (conf. 87) | −2421.018446 | 1.71 | 1 | 1.77 | 2 |
| 6 (conf. 90) | −2421.019218 | 1.23 | 2 | 0.00 | 32 |
| 7 (conf. 1) | −2344.795749 | 0.50 | 22 | 0.99 | 10 |
| 7 (conf. 2) | −2344.796541 | 0.00 | 51 | 0.00 | 52 |
| 7 (conf. 3) | −2344.793589 | 1.85 | 2 | 2.20 | — |
| 7 (conf. 4) | −2344.795708 | 0.52 | 21 | 0.93 | 11 |
| 7 (conf. 6) | −2344.793915 | 1.65 | 4 | 0.39 | 27 |
| Amidea | αb | β1c | β2c | β3c | γ1d | γ2d | γ3d | ωe | l1f | l2g | l3h | l4i | l5j | Helicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Conformers are numbered according to their appearance during conformational search.b α = H–C*–N–C(O).c β = OC–C–Cipso.d γ = C(O)–C–Cipso–Cortho (of the two possibilities the absolute values ≤90° has been chosen).e ω = N–C*–C*–N.f l1 = (C)O⋯H(C*).g l2 = (C)O⋯H(Cortho).h l3 = (N)H⋯Cipso.i l4 = Csp3(trityl)⋯Csp3(trityl).j l5 = (C)O⋯HN.k C2-symmetry conformer.l α = C3–C2–N–C(O).m ω = C1–C2–C2′–C1′.n l1 = (C)O⋯H(C6).o l5 = C2⋯HN. |
||||||||||||||
| 1A (X-ray) | 2.4 | −83.8(3) | 156.1(2) | 33.9(3) | −26.4(3) | −50.8(3) | −57.6(3) | −117.7(2) | 2.438 | 2.407 | 2.336 | 6.913(3) | MMM | |
| −8.5 | −99.3(3) | 138.6(2) | 16.6(3) | −2.1(3) | −54.0(3) | −56.3(3) | 2.446 | 2.473 | 2.359 | OMM | ||||
| 1B (X-ray) | 38.8 | −65.9(3) | 52.9(3) | 173.8(2) | 22.3(3) | 48.7(3) | 64.5(3) | −90.1(2) | 2.471 | 2.260 | 2.424 | 6.975(3) | PPP | |
| 2.6 | −85.1(3) | 155.2(2) | 33.3(3) | −29.3(3) | −52.1(3) | −58.8 | 2.420 | 2.419 | 2.362 | MMM | ||||
| 1 (conf. 1) | −35.6 | −99.7 | 139.4 | 18.5 | −23.4 | −52.9 | −58.8 | −72.7 | 2.551 | 2.467 | 2.394 | 6.472 | 2.158 | MMM |
| 54.7 | −103.3 | 13.6 | 134.7 | −5.3 | −55.0 | −60.0 | 2.682 | 2.415 | 2.533 | OMM | ||||
| 4a (X-ray) | −11.7 | 80.3(2) | −159.2(2) | −35.9(2) | 15.3(2) | 60.3(2) | 61.1(2) | −65.5(2) | 2.448 | 2.423 | 2.301 | 5.486(3) | PPP | |
| 16.9 | 61.2(2) | −55.1(2) | −178.2(2) | 34.4(2) | 50.5(2) | 64.2(2) | 2.398 | 2.220 | 2.311 | PPP | ||||
| 4a (conf. 3) | −17.1 | 84.2 | −155.0 | −32.5 | 21.0 | 56.2 | 59.5 | −60.0 | 2.373 | 2.400 | 2.310 | 6.012 | 2.777 | PPP |
| 31.1 | 48.9 | −68.3 | 169.1 | 39.2 | 39.2 | 68.0 | 2.367 | 2.162 | 2.379 | PPP | ||||
| 4b (X-ray) | −8.8 | −113.0(2) | 120.9(1) | 3.1(2) | −5.0(2) | −39.2(2) | −69.9(2) | −69.2(2) | 2.302 | 2.824 | 6.166(2) | OMM | ||
| −4.4 | −115.4(2) | 119.6(2) | 1.4(2) | 2.6(2) | −46.0(2) | −65.9(2) | 2.266 | 2.866 | OMM | |||||
| 4b (conf. 8)k | −174.0 | 123.1 | −110.9 | 7.1 | −3.2 | 53.0 | 57.4 | −46.1 | 3.772 | 2.615 | 6.707 | OPP | ||
| 5 (X-ray) | −171.3 | 123.0(1) | 4.8(2) | −115.8(1) | −5.7(2) | 50.3(2) | 68.1(2) | 87.4(1) | 3.785 | 2.470 | 2.647 | 6.868(2) | 2.184 | MPP |
| 29.4 | −123.5(1) | −6.4(2) | 112.2(2) | 20.6(2) | −42.8(2) | −68.5(1) | 2.365 | 2.416 | 2.755 | PMM | ||||
| 5 (conf. 60) | 26.0 | −126.6 | −8.4 | 111.9 | 13.2 | −49.0 | −68.7 | 87.0 | 2.349 | 2.474 | 2.697 | 6.713 | 1.918 | PMM |
| −168.5 | 148.3 | 30.5 | −89.0 | −35.5 | 37.6 | 77.3 | 3.893 | 2.485 | 2.442 | MPP | ||||
| 6 (X-ray) | −9.4 | −91.7(2) | 25.8(2) | 147.8(2) | 30.0(2) | 43.0(2) | 75.7(2) | −104.3(2) | 2.364 | 2.443 | 2.464 | 6.691(2) | PPP | |
| 39.2 | 53.3(2) | −63.4(2) | 174.1(2) | 42.9(2) | 48.7(2) | 66.7(2) | 2.479 | 2.296 | 2.345 | PPP | ||||
| 6 (conf. 90)k | 35.2 | −62.6 | 55.6 | 177.5 | −36.9 | −39.6 | −64.8 | −104.9 | 2.418 | 2.125 | 2.301 | 7.392 | MMM | |
| 7 (conf. 2) | −9.3l | 86.7 | −151.8 | −29.7 | 22.0 | 53.4 | 59.4 | −92.1m | 2.139n | 2.397 | 2.327 | 6.882 | 2.380o | PPP |
| −19.7l | 34.2 | 151.3 | −85.3 | 21.8 | −45.6 | 89.2 | 2.225n | 2.293 | 2.430 | 2.369o | PMP | |||
 | ||
| Fig. 2 Calculated structures of the lowest-energy conformers of triphenylacetamides 1 (a), 4a (b), 4b (c) and 5 (d). Dashed lines indicate possible attractive interactions. Geometrical parameters describing these interactions are listed in Table 2 in text. Some hydrogen atoms were omitted for clarity. | ||
Computationally found number of thermally accessible conformers was lower than reported previously for N,N′-ditrityldiamines or O,O′-ditrityldiols.6 Only in the case of amide 6 the number of the individual low-energy structures exceeded seven. Comparison of energetic and structural data let us to a few conclusions. Statistically, the preferred conformation of the molecule associated with the α torsion angles was either syn or gauche. The parallel or close to parallel orientation of CO and C*–H dipoles allowed existence of intramolecular C*H⋯OC hydrogen bonds closing the five membered ring and a decrease of the total energy of the molecule. However in some cases, i.e. in the lowest energy conformer of 4b, the value of the α angle was close to 180°, indicating anti orientation of the CO and C*–H dipoles, while in the crystals of 4b the syn orientation remained. The lowest-energy conformer no 8 of 4b is characterized by the stabilizing C*H⋯OC hydrogen bonds formed between the protons attached to the distal stereogenic centers and the carbonyl oxygen atoms, thus establishing communication between the two centers. Most probably, the energetic benefit connected with the formation of the intramolecular hydrogen bonds closing the six-membered rather than five-membered ring has balanced an increase of energy due to the steric crowding caused by N-methyl groups. Similar situation was observed for 5, although in this particular case, the anti-conformation associated with one of the α angles was a result of formation of intramolecular hydrogen bond of the NH⋯OC type, an unprecedented situation that has also been reflected in crystals of 5.
Bisamide 7 was the special case since chirality of the reporter units was triggered by chiral axis instead of the stereogenic center. We have found that the values of the α angles (defined here as C3–C2–N–C(O) using the standard numbering of carbon atoms in biphenyl framework) indicated the synperiplanar (sp) conformation for all structures, regardless of their relative energies. This orientation allowed formation of (C3)H⋯OC hydrogen bonding and lead to the parallel orientation of the N–H bonds with respect to the C1–C1′ bond. The amide moieties were lying coplanar with the respective aromatic rings of the chiral inductor.
In crystals, in all but one structure the absolute values of the α angles indicate sp orientation of the nearest C*–H and OC bonds. The exception was diamide 5, in which, similarly as in the calculated lowest-energy structure, the adoption of the anti conformation allowed formation of the intramolecular hydrogen bond of the NH⋯OC type, unique in this series of structures. In crystals, deviations from the sp conformation are substantial, the biggest being invariably associated with the PPP helicity of the Tr groups. This might suggest that the PPP helicity is less suited to be stabilized by the intramolecular C*–H⋯OC hydrogen bonds closing the five membered rings.
As it was mentioned previously the calculated lowest-energy conformer no 8 of 4b is characterized by anti orientation of the C*–H and OC bonds and OPP helicity of the Tr groups, while in the crystal the sp conformation described by the α angle is combined with OMM helicity of the Tr group, a clear illustration of the flow of information from the stereogenic center to the Tr group directly associated with this center.
Conformation defined by a set of β angles depends on the nature of the amide (secondary vs. tertiary). In the lowest energy conformers of secondary aliphatic amides 1 and 4a, the carbonyl group is oriented either sp or synclinally (sc) with respect to one phenyl ring of the trityl moiety. The second and the third β angles are correlated with this β angle and assume either anticlinal (ac) orientations (if the first one was sp (or sc)) or anti, if the first β angle assumed the sc conformation. The same pattern of β angles was found for the higher energy conformers of 1 and 4a. When methyl groups were introduced at the nitrogen atoms, as in tertiary amide 4b, the carbonyl groups became sp (β1 value approximated 0°) and the amide methyl group ap with respect to one of the phenyl rings, and in bisecting position with respect to the remaining two phenyl substituents. Situation was more complicated for amides 5 and 6 having aromatic substituents. Analysis of data from Tables 1 and 2 led to the conclusion that “conformational behavior” of 5 was closer to the tertiary amide 4b than to its secondary equivalent 4a. In the lowest energy conformer no 60 of amide 5 the conformation described by a set of β1–β3 angles could be described as sp/ac/ac and sc/ac/ac while for the lowest-energy conformer of 6 as sc/sc/ap. This diversity was found also for the low-energy conformers of 7. For the lowest-energy conformer no 2 of 7 the conformation described by a set of β angles can be described as sp(sc)/ac/ac. In chiral bis(triphenylacetamides) the structure of the reporter unit approximated to a distorted C3 symmetry, both in the gas phase and in majority of crystals. At the first glance there was no simple model of helicity induction in this series. For example, in both 1 and 4a the absolute configuration of the sterogenic centers was R, but helicities of the reporter units were different. Considering only the lowest energy conformers of 1 and 4a, the overall chirality of the most energetically preferred diastereomers could be described as (R,R,M,M,M,O,M,M) and (R,R,P,P,P,P,P,P), respectively. Results of the theoretical calculations indicated that introduction of methyl groups at the nitrogen atoms in 4b did not substantially affect the conformation of the reporter units: for the lowest-energy and C2-symmetrical conformer no 8, the helicity of the trityl groups remained OPP. However, in crystals methylation lead to the OMM conformer. The case of 5 was exceptional. Apart from the C2-symmetrical conformers, the remaining structures were characterized by the formal object to mirror-image relationship between the trityl groups within the same molecule. Also in the crystals of 5 the two Tr fragments mutually adjusted their conformation in opposite sense (i.e. PMM and MPP) to provide a molecule in which orientation of the whole tritylamide fragments is mutually opposite (all characteristic torsion angles, listed in Table 2, are of the opposite sign). This change is accompanied by a change in sign of the ω angle which becomes positive, while in all other conformers its sign is always negative.
The cases of 6 and 7 were more consistent. In majority of low-energy conformers of 6 and 7, as well as in crystals of 6 the elements of chirality in the parent amine skeletons induced homohelicity in the reporter units (i.e. all the γ angles within the same group had the same sense of twist). The special case was the conformer no 1 of amide 6, where indeed each of the trityl groups was homohelical, but their mutual relationship was like object to its mirror image.
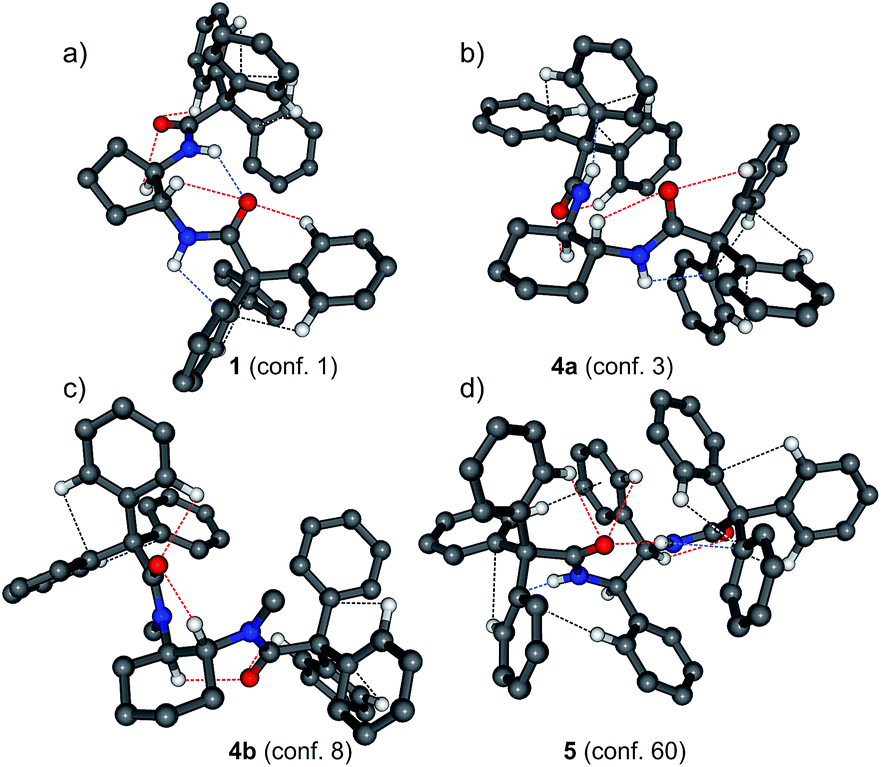
Analysis of the X-ray results leads to the general conclusion that in enantiomerically pure crystals of 1, 4a, 4b and 6 the Tr groups display a propeller-like conformation, which becomes significantly deformed upon methylation (4b). In crystals build of molecules of the same chirality, the propeller shaped Tr groups exhibit either MMM or PPP type of helicity. However, in one molecule the two propellers are usually of the same helicity. Presumably this assures the lack of steric conflict between the two rotors. Only in one of the five cases a pair of Tr groups in one molecule displays opposite helicity (amide 1, molecule B, Table 2). This modification seems necessary for the carbonyl oxygen atoms to get involved in many, relatively strong intermolecular C–H⋯O interactions (Table S3†) as opposed to the corresponding, much weaker interactions displayed by molecule A. This is clearly illustrated in Fig. 3, which is a collection of 2D fingerprint plots of Hirshfeld surfaces calculated for the molecules of 1A and 1B with the use of program Crystal Explorer.24 These plots (plot of di versus de, where de and di are the distances to the nearest atom center exterior and interior to the molecular surface) show that there is a distinct difference between the two symmetry independent molecules, 1A and 1B, in terms of C–H⋯O interactions. This is reflected in appearance of characteristic strips at the left side of the plot for the molecule B that are absent in the plot for the molecule A. The same concerns the C–H⋯π interactions that are represented by the characteristic wings that appear in a plot for the molecule B at lower values of de and di than in a plot for the molecule A. Hence, it seems as though a pair of Tr groups that exhibits the opposite sense of helicity in one molecule is advantageous form the point of view of maximization of intermolecular interactions. The inversion of a sense of rotation of one of the propellers, which takes place in the molecule B, is accompanied by another conformational changes, e.g. a substantial change in a value of the ω angle which controls mutual orientation of the two tritylamide moieties. In the molecule A, having both Tr groups of the same helicity, the two groups are in (−)ac orientation (ω = −117.7(2)°), while in the molecule B, in which the two Tr groups have opposite helicity, they are in (−)sc orientation (ω = −90.1(2)°).
 | ||
| Fig. 3 Full fingerprint plots (FPs) for two independent molecules in crystals of 1 and fingerprint plots resolved into H⋯C and H⋯O close contacts. The first row is related to molecule A and the second row to molecule B. The corresponding Hirshfeld surface plots are provided in the ESI (Fig. S2†). | ||
Detailed analysis of the X-ray results reveals that deformation of the propeller shape in crystals is always unidirectional, i.e. leads to the OMM conformer. Such conformer seems to generate more space around, the otherwise completely shielded, nitrogen atom. For example, in the amide 1 the number of short intramolecular contacts involving the NH group is higher in the moieties that contain Tr groups of either MMM or PPP helicity than in a moiety that contains the Tr group of the OMM helicity (see Table S2, ESI†). Such a demand for more space around the amide nitrogen arises upon introduction of methyl substituent at the nitrogen atom, as in the case of 4b, in which both Tr groups have OMM helicity in crystal, while 4a contains both the Tr groups of PPP helicity.
According to the experimental and computational data, the NH⋯O(C) hydrogen bonding is only partly responsible for the control of conformation of amides under study. In the majority of conformers of 1, 4a, 4b, 6 and 7 the amide hydrogen bonding does not exist. This is caused by the rigidity of the carbon skeleton to which the amide groups are attached and shielding effect of hydrophobic trityl groups.
For the lowest-energy conformer no 1 of amide 1 the calculated NH⋯O(C) distance amounts to 2.158 Å and one can say that in this case the hydrogen bond controls the molecular conformation. On the other hand, for 4a having rigid cyclohexane skeleton, the shortening of the NH⋯O(C) distance did not result in decrease of the total energy of the conformer. The NH⋯O(C) distance (2.777 Å) calculated for the lowest-energy conformer no 3 was over 0.6 Å longer than that calculated for the higher in energy conformer no 1. The shortest NH⋯O(C) distance within all calculated individual structures of amides under study was found for the lowest-energy conformer no 60 of the amide 5. The calculated H⋯A distance amounted to 1.918 Å whereas the value obtained from X-ray study of the crystals of 5 was longer and measured 2.18 Å (Table S2, ESI†). In other words – the amide hydrogen bonding in this type of compounds constitutes rather an exception than a real structure-determining factor.
Detailed look at the data collected in Table 2 reveals correlation between l4 (the distance between the trityl groups within the same molecule) and the value of the α angle. High values of the α angle describing the ap conformation are associated with the longest distances between the ipso carbon atoms.
Packing of the molecules in crystals
Hydrogen-bond parameters are given in Tables S2 (intramolecular) and S3 (intermolecular) in ESI.† A concise summary of the intermolecular interactions occurring in the crystals of 1, 4a, 4b, 5 and 6 is provided in Table S4,† which lists the relative contributions to the Hirshfeld surface areas of the molecules 1, 4a, 4b, 5 and 6 of the various intermolecular contacts (H⋯H, H⋯C, H⋯O, C⋯C, H⋯N, C⋯Cl and H⋯Cl).24 Molecules that have the Tr groups attached to the hydrocarbon rings (1, 4a, 4b and 6) display packing difficulties which are manifested either by duplication of the content of the asymmetric unit (Z′ = 2) as in 1 or by incorporation of CH2Cl2 solvent molecules into the crystal structures of 4a and 4b, or by the presence of microporosities, as in the crystals of 4b and 6. Crystals of 5, in which the Tr groups attached to a carbon chain adopt mirror related conformations, are free from the above mentioned obstacles. The two independent molecules in the crystals of 1 differ in conformation and in the involvement of the molecules in intermolecular interactions (vide supra).Compounds 4a and 4b form dichloromethane solvated crystals. Two types of recognition element are independently involved in the guest binding. The first is the hydrogen bond accepting carbonyl groups, which interact with several binding sites inside the cage associated with the set of CH groups, including those of dichloromethane. The second recognition element is the dichloromethane chlorine atoms, which act as the hydrogen bond acceptors from the trityl CH groups. This has been illustrated on the fingerprint plots, constructed independently for the host and the guest molecules, which are shown in Fig. 4. The fingerprint plots are quite asymmetric; this is expected, since interactions occur between two different species (host and guest). The solute and solvates show spikes arising from the weak interactions of the C–H⋯Cl and C–H⋯O type. In each case the upper spike (de > di) corresponds to the hydrogen bond donor and the lower spike (de < di) to the hydrogen bond acceptor.
 | ||
| Fig. 4 Fingerprint plots for solute (first column) and dimethylchloride solvent molecules (second column) in the crystals of 4a and 4b resolved into H⋯Cl (first and third row) and H⋯O (second and fourth row) close contacts. The corresponding Hirshfeld surface plots are provided in the ESI (Fig. S2†). | ||
For 4a, the spike arising from H⋯O contacts is sharp due to very short H⋯O distance (Table S3†). This interaction is weaker in 4b (higher de and di values). Exactly opposite relationship concerns the spikes arising from C–H⋯Cl interactions in 4a and 4b.
Full fingerprint plots derived from Hirshfeld surfaces for single component crystals of 5 and 6 (Fig. S2 of ESI†) reveal in both structures an increase in number of the C–H⋯π interactions and in their strength due to the presence of additional π-electron groups in the inductor skeleton. Additionally, in the fingerprint plot of 6 there is a large number of points at high values of de and di, which extend beyond the plotted range. This points to the presence of voids in the crystal structure of 6. The voids occupy 0.8% of the unit cell volume, as estimated using a probe vector of 1.2 Å (ref. 23) and are situated in between a nearly parallel phenyl rings belonging to the inductor and reporter groups of the same molecule (Fig. 5), so in this context the voids should rather be ascribed as intramolecular. The free space created at one side of the molecule is contrasted with relatively compact arrangement of atoms at the opposite side, where we observe very short intramolecular C–H⋯π interaction between the trityl groups (see Fig. 5 and Table S2† for geometrical details).
 | ||
| Fig. 5 Space-filling representation of molecule 6 present in crystal. Free space in between phenyl rings belonging to the inductor and reporter groups is marked in light-brown color. Atoms involved in strong C–H⋯π interaction between trityl groups are marked in light blue color. | ||
Characteristic feature of the investigated crystals is the complete hindrance of the amide N–H groups and their inability to get involved in any intermolecular interactions as well as their highly restricted ability for formation of intramolecular hydrogen bonds. This is due to the presence of the Tr groups, which act as supramolecular N–H protecting groups, especially when attached to the rigid carbocycles. The compounds having the amide groups that form strong intermolecular hydrogen bonds are characterized by low solubility in many organic solvents. Here we observe the enhanced solubility of the whole group of investigated compounds compared to reagents with smaller substituents.
Electronic circular dichroism studies
The electronic circular dichroism (ECD) spectroscopy was chosen as a method of choice for studying the chirality transfer within bis(trifenylacetamides) 1–8.25–27 The comparison of experimentally measured ECD spectra with those calculated for the low energy conformers of selected amides 1, 4–7 allowed confirmation of the correctness of the above-mentioned structural studies. Examples of the ECD spectra measured and calculated for arbitrarily chosen amides 1, 4a–4b, 5, 6 and 7 are shown in Fig. 6 (see ESI† for the remaining results). For clearer discussion the amides may be divided into three groups due to the differences in their structures. The first group consisted of “pure” aliphatic amides 1 and 4. To the second group belonged amides 2, 3, 5 and 6, bearing additional π-electron substituent. The last group constituted from the axially chiral aromatic amides 7 and 8. | ||
| Fig. 6 Examples of experimental (solid black lines) and calculated ΔE-(solid blue lines) and ΔΔG°-Boltzmann averaged (dashed blue lines) ECD spectra of triphenylacetamides 1 and 4, 5 and 7. The calculated ECD spectra were wavelength corrected to match experimental absorption bands. | ||
The set of bis(triphenylacetamides) (1–8) prepared for this study, was characterized by R absolute configuration of elements of chirality and displayed characteristic Cotton effects (CE) in their ECD spectra within the 250–185 nm range (see Table 3 and full version of it in ESI†). The secondary aliphatic derivatives 1 and 4a, with R configuration at the stereogenic centers exhibited a negative Cotton effect within the 1Lb transition range (ca. 220–210 nm) and a positive CD couplet within the strong 1B band (200–195 nm). The sequence of signs of Cotton effects was reversed and the respective absorption bands were significantly enhanced and red-shifted in the case of 4b, when amide hydrogen atoms were replaced by methyl groups. This was a result of a substantial helicity change in the reporter unit after N-methylation.
| Amide | Δε (nm) | ε (nm) | |
|---|---|---|---|
| 1 | −19.0 (219) | 29.2 (196) | 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (192) 000 (192) |
| 2 | 31.6 (213) | 29.2 (196) | 209400 (189) |
| 3 | −19.6 (213) | −40.3 (197) | 150600 (192) |
| 4a | −18.5 (223) | 24.9 (198) | 148600 (194) |
| 4b | 45.7 (232) | −97.9 (211) | 142900 (192) |
| 5 | −6.5 (239) | −66.5 (208) | 208500 (190) |
| 6 | −126.5 (212) | 212.1 (193) | 204300 (189) |
| 7 | 6.3 (227) | −30.5 (212) | |
| 65.1 (196) | 170900 (194) |
||
| 8 | −67.6 (219) | 160.0 (201) | 159400 (194) |
The presence of π-electron group in the inductor skeleton had contrasting effects on the ECD spectra. In the case of 3, double bond in cyclohexene ring did not affect the signs of the Cotton effects. When compared to 4a, the short-wavelength CE measured for 3 showed small hyperchromic effect as a result of summation of absorption within trityl and double bond chromophores. The strong hyperchromic effect was observed for amides 5 and 6, whereas the highest amplitude has been found for the amide 6. For both 5 and 6 the sequence of signs of Cotton effects: negative long-wavelength and positive short-wavelength resembled these found for aliphatic amides.
Due to the presence of conjugated π-systems, the measured ECD spectra for 7 and 8 showed very complex combination of positive and negative CE's especially in the low-energy region. Considering only the region of the trityl absorption (below 220 nm) the ECD spectrum of 7 was similar to that measured for other amides. The exception was the amide 8, the measured ECD spectrum was rather a result of exciton coupling between electric transitions dipoles polarized along the long axis of each naphthyl group than induced chirality in the trityl groups.26
Change of an environment polarity from cyclohexane to acetonitrile did not influence the pattern and had very little effect on magnitudes and transition energies of respective CE's. This indicated that both, the structure and the conformational equilibriums remained more or less the same in both media.
Depuzzling of the ECD spectra
A comparison between the experimental and the calculated ECD spectra for amides 1, 4–7 showed their almost perfect agreement. This way the correctness of the above-mentioned structural studies was confirmed. However, a detailed look into the calculated ECD spectra should allow us to determine the factors that are responsible for the observed CE's. We were particularly interested whether the observed ECD spectra result from intrachromophoric or interchromophoric interactions and wished to clarify to what extent the aromatic substituents affected the overall ECD spectrum of a given amide.To solve this issue we selected amides 1, 5 and 7 (Fig. 7) as representative of each of the earlier mentioned group. We considered only the lowest-energy conformers of these amides, since they were responsible mainly for observed and calculated ECD spectra (see Fig. A6 in ESI†).
 | ||
| Fig. 7 Model compounds used in the study. | ||
We started our study from conformer no 1 of amide 1. This was the simplest case since the calculated and observed Cotton effects came solely from the induced chirality within the trityls and the intra- and/or interchromophoric interactions. We divided (“cut”) the conformer into two moieties, denoted here as 1(s1) and 1(s2). Each of them consisted of cyclopentane ring and one of the two triphenylacetamide substituents. The second one was replaced by a simple acetamide fragment. Apart from the acetamide, the other structural parameters remained the same as in the parent structure. The moiety 1(s1) was characterized by MMM and the moiety 1(s2) by OMM helicity of the trityl chromophore.
For each of the sub-structures the ECD spectrum was calculated at the M06-2X/6-311G(2d,2p) level (see Fig. 8a and A7 in ESI†). Both spectra showed the same sequence of CE's – the long-wavelength 1Lb negative and positive 1B effect in the higher energy region. Surprisingly, the magnitudes of CE's calculated for C3-symmetrical chromophore were smaller than those for 1(s2) characterized by C1-symmetry of the reporter unit. Finally, the calculated ECD spectra for 1(s1) and 1(s2) were summed up and the resulting spectrum was compared with those calculated for the lowest energy conformer no 1. The difference between respective rotatory strengths depended on the type and energy of electronic transition. Calculated for conformer no 1, the rotatory strength appeared in lower energy region assumed 83 × 10−40 erg esu cm Gauss−1 and was lower than those for summed up spectrum (110 × 10−40 erg esu cm Gauss−1). The higher energy region was more consistent and the difference between respective rotatory strengths was lower than 6%. This suggests that the interchromophoric interactions were of minor importance, and both reporter units gave independent contribution to the overall ECD spectrum.
 | ||
| Fig. 8 Calculated ECD spectra for model compounds: left column: (a) 1(s1) (black line) and 1(s2) (red line); (b) 5(s1) (black line) and 5(s2) (red line); (c) 7(s1) (black line) and 7(s2) (red line); right column: (d) the sum of 1(s1) and 1(s2) (blue line) and conformer 1 of amide 1 (black dashed line); (e) the sum of 5(s1) and 5(s2) (blue line) and conformer 60 of amide 5 (black dashed line); (f) the sum of 7(s1) and 7(s2) (blue line) and conformer 2 of amide 7 (black dashed line). Wavelengths were not corrected; vertical bars represented calculated rotatory strengths. | ||
The conformer no 60 of amide 5 represented more complex system due to the presence of phenyl substituents in the parent amine skeleton. The inductor may generate intrinsic ECD signal in the same spectral region as the trityl chromophore and the observed and calculated ECD spectra for 5 may result from overlapping of the induced and intrinsic effects. Similarly to the previously discussed example, the structure of the conformer no 60 was cut up several sub-parts, the most important were N,N′-diacetyl-1,2-diaminodiphenylethane (5(s1), “the inductor”) and N,N′-bis(triphenylacetyl)-1,2-diaminoethane – (5(s2), “the reporter”, Fig. 7). All the structural parameters characterizing these structures (angles, distances) were the same as in the parent conformer.
The calculated ECD spectra for model compounds were shown in Fig. 8b (see also Fig. A8 in ESI†). As we expected, the inductor generated intrinsic rotatory strengths of the same sequence negative/positive of CE's as calculated for the parent structure. Calculated for 5(s1) energies of respective transitions were blue shifted and magnitudes were significantly lower than calculated for conformer no 60 of 5. The substructure 5(s2) consisted of two chromophores being in the formal relation like an object to its mirror-image. However, the calculated rotatory strengths followed general order – negative long wavelength and positive short wavelength CE's. The calculated rotatory strength magnitudes for 5(s2) were 4–5 times higher than those calculated for 5(s1). The comparison of summed up ECD spectrum with calculated one for the conformer no 60 of amide 5 showed their excellent agreement. To generalize, for amide 5 the induced CE's within the reporter units overlapped intrinsic CE's.
The amide 7 represented the most complex system. The inherently chiral and configurationally stable biphenyl moiety may dominate the overall ECD spectrum. Repeating the simplification procedure we divided the lowest energy conformer no 2 of amide 7 into two main parts, where 7(s1) had the biphenyl fragment and 7(s2) consisted of two N-methyltriphenylacetamides (Fig. 7). The calculated for 7(s1) ECD spectrum, shown in Fig. 8c (see also Fig. A9 in ESI†) exhibited very small rotatory strengths in the higher energy region. In this particular spectral region the biphenyl chromophore was “silent” and generated no CE's. This was because both “halves” of 7(s1) were twisted almost perpendicularly and there was no conjunction between π-clouds of the two parts of the molecule.26,27 The comparison of the ECD spectra calculated and summed up for the conformer no 2 of 7 and the sub-unit 7(s2) confirmed unambiguously that in the spectral region, typical for electronic absorptions within the trityl chromophore, the overall ECD spectrum of 7 resulted from the CE's induced within each of the reporter units.
Conclusions
We have observed the chiral transmission in the rationally designed bis(triphenylacetamides). With reference to the questions raised in the Introduction, we were able to demonstrate that for the secondary bis(triphenylacetamides) there is a general correlation between the absolute configuration of the element of chirality and observed chiroptical response (Fig. 9). | ||
| Fig. 9 Model of the optical activity of bis(triphenylacetamides). | ||
For all experimental and computed ECD spectra the uniformly identical sequence of signs of Cotton effects had been found: negative at around 215 nm and positive below 200 nm, even though the conformer structures and conformer populations were different. This result can only be explained by the dependence of the ECD spectra on the absolute configuration of the proximal stereogenic element, which was uniformly R. Note, that for O,O′- and N,N′-ditrityl derivatives of the same absolute configuration the sequence of the respective CE's was opposite.6
The model of optical activity proposed here for the bisamides is in good agreement with that proposed previously for monoamides.7 The observed and calculated ECD spectra resulted from independently induced chirality within each of the reporter units. Even for compounds having phenyl groups and/or possessing the plane chirality, the intrinsic CE's gave only negligible contribution to the overall ECD spectra.
The trityl⋯trityl interactions gave only minor contribution to the chirality induction within the molecule. For ditrityl ethers and amines the structural feature that served to identify mutual interactions of the trityl groups was their conformation – the trityls that were not involved in steric interactions had an approximate C3-symmety. However, this concerned derivatives with distances between the ipso carbon atoms in a range from 5.1 to 5.4 Å.6 Meanwhile, introduction of an amide linker between the inductor and the receptor caused a significant increase of this distance, which resulted in consequent lack of involvement of the two reporter units in mutual steric interactions. As a result, the trityl groups in bis(trifenylacetamides) are C3 symmetrical (or nearly so). Therefore, the macroscopic equivalent of bisamides is rather a double-rotor helicopter than a bevel gear.
The results reported so far were focused on specific type of induction – from the point stereogenic center to conformationaly labile trityl chromophore. For the first time we have shown that for the effective chirality transmission the presence of the stereogenic center was not mandatory. Compounds having chirality axis may act as the effective helicity inductors as well.
As an additional outcome of our study comes the evidence that the trityl group, commonly used in organic synthesis as a protecting group, can also play an analogous role in supramolecular chemistry, where it prevents formation of hydrogen bonded amide⋯amide supramolecular synthons.
Acknowledgements
This work was supported by research grant from National Science Center (NCN) Poland no UMO-2011/03/ST5/01011. All calculations were performed in Poznan Supercomputing and Networking Center (grant no. 217).Notes and references
- (a) P. J. Kocienski, Protecting Groups, Thieme, 2005, pp. 269–274 Search PubMed; (b) T. W. Greene and P. G. M. Wats, Protective Groups in Organic Synthesis, Wiley, New York, 2006, pp. 152–156 Search PubMed.
- (a) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348 CrossRef CAS; (b) W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25 CrossRef CAS PubMed.
- (a) A. Khazaeia, M. A. Zolfigol, A. R. Moosavi-Zare, A. Zare, A. Parhami and A. Khalafi-Nezhad, Appl. Catal., A, 2010, 386, 179 CrossRef PubMed; (b) A. Khazaei, M. A. Zolfigol, A. R. Moosavi-Zare, F. Abi, A. Zare, H. Kaveh, V. Khakyzadeh, M. Kazem-Rostami, A. Parhami and H. Torabi-Monfared, Tetrahedron, 2013, 69, 212 Search PubMed; (c) A. Khazaei, M. A. Zolfigol, A. R. Moosavi-Zare, A. Zare, M. Khojasteh, Z. Asgari, V. Khakyzadeh and A. Khalafi-Nezhad, Catal. Commun., 2012, 20, 54 CrossRef CAS PubMed; (d) J. Bah and J. Franzén, Chem.–Eur. J., 2014, 20, 1066 CrossRef CAS PubMed; (e) O. Bassas, J. Huuskonen, K. Rissanen and A. M. P. Koskinen, Eur. J. Org. Chem., 2009, 1340 CrossRef CAS PubMed; (f) L. Busetto, M. C. Cassani, C. Femoni, M. Mancinelli, A. Mazzanti, R. Mazzoni and G. Solinas, Organometallics, 2011, 30, 5258 CrossRef CAS.
- J. Ściebura, P. Skowronek and J. Gawronski, Angew. Chem., Int. Ed., 2009, 48, 7069 (Angew. Chem., 2009, 121, 7203) CrossRef.
- (a) N. Tohnai, Y. Mizobe, M. Doi, S. Sukata, T. Hinoue, T. Yuge, I. Hisaki, Y. Matsukawa and M. Miyata, Angew. Chem., Int. Ed., 2007, 46, 2220 CrossRef CAS PubMed; (b) N. Veldman, A. L. Spek, J. J. H. Schlotter, J. W. Zwikker and L. W. Jenneskens, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 174 CrossRef; (c) R. Destro, T. Pilati and M. Simonetta, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2495 CrossRef; (d) B. Kahr and R. L. Carter, Mol. Cryst. Liq. Cryst., 1992, 219, 79 CrossRef CAS PubMed.
- J. Sciebura, A. Janiak, A. Stasiowska, J. Grajewski, K. Gawronska, U. Rychlewska and J. Gawronski, ChemPhysChem, 2014, 15, 1653 Search PubMed.
- N. Prusinowska, W. Bendzińska-Berus, M. Jelecki, U. Rychlewska and M. Kwit, Eur. J. Org. Chem., 2015, 738 CrossRef CAS PubMed.
- (a) V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, J. Am. Chem. Soc., 2001, 123, 2979 CrossRef PubMed; (b) J. Gawronski, M. Kwit and K. Gawronska, Org. Lett., 2002, 4, 4185 CrossRef CAS PubMed.
- Scigress 2.5, Fujitsu Ltd Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Grimme and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 12639 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 Search PubMed; (c) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS; (d) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (b) D. Jacquemin, E. A. Perpète, I. Ciofini, C. Adamo, R. Valero, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2071 CrossRef CAS.
- (a) S. Ehrlich, J. Moellmann and S. Grimme, Acc. Chem. Res., 2013, 46, 916 CrossRef CAS PubMed; (b) T. Risthaus and S. Grimme, J. Chem. Theory Comput., 2013, 9, 1580 CrossRef CAS; (c) J. Moellmann and S. Grimme, J. Phys. Chem. C, 2014, 118, 7615 Search PubMed.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS PubMed.
- (a) S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed; (b) L. Goerigk and S. Grimme, J. Phys. Chem. A, 2009, 113, 767 CrossRef CAS PubMed.
- (a) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS PubMed; (b) Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425 Search PubMed.
- N. Harada and P. Stephens, Chirality, 2010, 22, 229 Search PubMed.
- M. Kwit, M. D. Rozwadowska, J. Gawronski and A. Grajewska, J. Org. Chem., 2009, 74, 8051 Search PubMed and references therein.
- CrysAlisPro, version 1.171, Agilent Technologies, Oxfordshire, England, 2013 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef.
- (a) J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef PubMed; (b) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC; (c) J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814 RSC; (d) F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- (a) C. Wolf and K. W. Bentley, Chem. Soc. Rev., 2013, 42, 5408 RSC; (b) N. Berova, L. diBari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914 RSC.
- Comprehensive Chiroptical Spectroscopy, ed. N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, Wiley, New York, 2012 Search PubMed.
- D. A. Lightner and J. E. Gurst, Organic Conformational Analysis and Stereochemistry from Circular Dichroism Spectroscopy, Wiley-VCH, New York, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full version of Tables 2 and 3, experimental details, copies of 1H and 13C NMR spectra, Cartesian coordinates for all calculated compounds, Tables A1–S6, Fig. A1–H6. CCDC 1414748–1414752. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra15554a |
| This journal is © The Royal Society of Chemistry 2015 |