
Acid-amplifying polymers: synthesis, characterization, and application to environmentally stable chemical amplification positive (ESCAP) resists†
Abstract
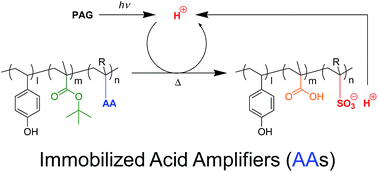
Acid amplifiers that are decomposed autocatalytically by acid species photogenerated from photoacid generators were synthesized and characterized. Such acid proliferation reactions by acid amplifiers were integrated into chemical amplification resists to improve the photosensitivity. In this study, new polymers bearing acid-amplifying (AA) units in their side chains were designed to suppress excess diffusion of acid species, and then evaluated for their photosensitivity with photoacid generators as environmentally stable chemical amplification positive (ESCAP) resists. The AA polymers have high photosensitivity (up to 1.4 mJ cm−2) after 254 nm of light irradiation and post-exposure baking at 140 °C. Using films of these polymers, 1 × 4 μm line-and-space patterns were fabricated.
 Please wait while we load your content...
Please wait while we load your content...

