DOI:
10.1039/C5RA15397B
(Paper)
RSC Adv., 2015,
5, 81689-81695
Solvent-controlled formation of four Ni(II) coordination polymers based on a flexible bis(imidazole) ligand: syntheses, structural diversification, properties†
Received
2nd August 2015
, Accepted 21st September 2015
First published on 21st September 2015
Abstract
To further investigate the influence of solvent in the self-assembly process, four new solvent-controlled isomeric Ni(II) coordination polymers based bib and 5-Br-H2ip ligands, namely, [Ni(bib)(5-Br-H2ip)(CH3CH2OH)]n (1), [Ni(bib)(5-Br-H2ip)]n (2), {[Ni2(bib)(5-Br-H2ip)2(H2O)]·0.5H2O}n (3), {[Ni(bib)(5-Br-H2ip)]·DMA}n (4) (bib = 1,4-bis(2-methylimidazol-1-yl)butane, 5-Br-H2ip = 5-bromoisophthalic acid) were prepared under the same reaction conditions except with diverse solvent systems. Their EA, IR, PXRD, single crystal XRD analysis and TG analysis have been characterized. As controlled different solvent systems, complex 1 exhibits an unusual [2 + 2] dia construction with the point symbol 66. Complex 2 shows a 3D non-interpenetrated dia structure with the point symbol 66. Complex 3 features a dinuclear 3D pillar-layer pcu network with the point symbol of 412·63. Complex 4 displays a 2D stacked layer sql net with the point symbol 44·62. The results demonstrate that solvent has a significant effect on the final structure of coordination polymers. Furthermore, the magnetic properties of complexes 1–4 have also been studied.
Introduction
During the past few years, there has been an enormous amount of attention on the design and fabrication of coordination polymers (CPs) owing to not only their diverse structural architectures and intriguing topological structures,1–3 but also promising applications in the fields of catalysis, magnetic properties, nonlinear optics, chemical sensors, gas storage and separation, and so on.4–8 Nevertheless, controlling the synthesis of CPs is a significant challenge as the self-assembly process can be manipulated by a great many external factors such as solvent, pH, temperature, concentration.9–12 Among these external stimuli, solvent is one of the most significant influence factors, which arises from their parameters such as polarity, boiling point, dielectric constant, van der Waals volume have an immense effect on the CPs synthesis process.13–15
Up to now, much effort has been devoted to research how the solvent affects the assemblies of coordination supramolecular systems. However, the mechanism of solvent effect is still poorly understood. Du et al. had commented how the solvent influence on the crystalline products from a kinetic or thermodynamic aspect.16 This can take place in following ways: (i) solvent as ligand;17 (ii) solvent as guest;18 (iii) solvent as both ligand and guest;19 (iv) structure-directing agent.20 Our group also utilized the flexible 1,4-bis(2-methylimidazol-1-yl)butane (bib) and the rigid 5-bromoisophthalic acid (5-Br-H2ip) exploring the solvent effect in our previous report.21 As is known to all, the flexible ligand is more sensitive to external stimuli, owing to their conformational changes.22–24 To further understand the role of solvent in the self-assembly process, bib and 5-Br-H2ip ligands continue to be employed to study this project.
In this contribution, four new solvent-controlled isomeric Ni(II) CPs based on bib and 5-Br-H2ip ligands, namely, [Ni(bib)(5-Br-H2ip)(CH3CH2OH)]n (1), [Ni(bib)(5-Br-H2ip)]n (2), {[Ni2(bib)(5-Br-H2ip)2(H2O)]·0.5H2O}n (3), {[Ni(bib)(5-Br-H2ip)]·DMA}n (4) were successfully synthesized under the same reaction conditions but different solvent media. Herein, their structural diversification, topology, thermal stability and magnetic properties have been investigated.
Experimental
Materials and general methods
All reagents and solvents employed were commercially available and used without further purification. Fourier transform infrared spectra were determined with a FT-IR 170 SX (Nicolet) spectrophotometer, and the samples were prepared as KBr pellets in the range 4000–400 cm−1. Elemental analyses for C, H, and N were performed with a Perkin-Elmer 2400C Elemental analyzer. Fluorescent spectra for the solid samples were measured with a Hitachi F-4500 fluorescence spectrophotometer at room temperature. Thermogravimetric analyses were carried out with a NETZSCH TG 209 thermal analyzer under a nitrogen atmosphere with a heating rate of 10 K min−1. Powder X-ray diffraction (PXRD) patterns were recorded on a Bruker D8 ADVANCE X-ray powder diffractometer (Cu-Kα, 1.5418 Å). Variable-temperature magnetic susceptibility data for polycrystalline 1–4 were obtained on a Quantum Design MPMS-XL-7 SQUID magnetometer under an applied field of 1 kOe over the temperature range of 2.0–300 K.
Synthesis of [Ni(bib)(5-Br-H2ip)(CH3CH2OH)]n (1)
A mixture of Ni(Ac)2·2H2O (24.9 mg, 0.10 mmol), 5-Br-H2ip (24.5 mg, 0.10 mmol), and bib (21.8 mg, 0.10 mmol) in EtOH (6 mL) was stirred under ambient conditions. The mixture was then placed into a Teflon-lined stainless steel (20 mL) container, heated at 150 °C for 3 days, and then cooled to room temperature at a rate of 5 °C min−1. After cooling to room temperature, green block crystals of 1 that were fit for single crystal X-ray were obtained in 42% yield. Anal. calcd for C22H27BrNiN4O5: C, 46.63; H, 4.77; N, 9.95. Found: C, 46.68; H, 4.81; N, 9.90. IR data (KBr, cm−1): 3445(m), 2940(w), 1678(w), 1548(s), 1450(s), 1379(s), 1283(w), 1156(m), 934(w), 861(w), 736(s), 678(s).
Synthesis of [Ni(bib)(5-Br-H2ip)]n (2)
Complex 2 was obtained by the same procedure used for preparation of 1 except that the EtOH was replaced by MeCN–EtOH mixed solvent (6 mL; v/v, 1/1). Green block crystals of 2 that were fit for single crystal X-ray were obtained in 39% yield. Anal. calcd for C20H21BrNiN4O4: C, 46.15; H, 4.04; N, 10.77. Found: C, 46.19; H, 4.07; N, 10.71. IR data (KBr, cm−1): 3367(w), 2923(w), 1837(w), 1593(s), 1482(m), 1378(s), 1288(m), 1164(m), 908(w), 778(m), 664(w).
Synthesis of {[Ni2(bib)(5-Br-H2ip)2(H2O)]·0.5H2O}n (3)
Complex 3 was obtained by the same procedure used for preparation of 1 except that the EtOH was replaced by MeCN–H2O mixed solvent (6 mL; v/v, 1/1). Green block crystals of 3 that were fit for single crystal X-ray were obtained in 51% yield. Anal. calcd for C56H54Br4Ni4N8O19: C, 39.65; H, 3.21; N, 6.60. Found: C, 39.79; H, 3.07; N, 6.81. IR data (KBr, cm−1): 3434(m), 1593(m), 1545(s), 1511(m), 1482(w), 1433(s), 1377(s), 1288(m), 1163(m), 1098(m), 778(s), 738(s), 683(w), 619(w).
Synthesis of {[Ni(bib)(5-Br-H2ip)]·DMA}n (4)
Complex 4 was obtained by the same procedure used for preparation of 1 except that the EtOH was replaced by DMA (6 mL). Green block crystals of 4 that were fit for single crystal X-ray were obtained in 65% yield. Anal. calcd for C24H29BrNiN5O5: C, 47.56; H, 4.82; N, 11.55. Found: C, 47.33; H, 4.76; N, 11.95. IR data (KBr, cm−1): 3429(m), 1648(m), 1595(s), 1547(s), 1507(w), 1441(s), 1376(s), 1290(w), 1118(m), 777(w), 740(m), 679(w), 619(m).
Crystallography
Single crystal X-ray diffraction analyses of complexes 1–4 were carried out on a Bruker SMART APEXII CCD diffractometer equipped with a graphite monochromated Mo-Kα radiation source (λ = 0.71073 Å) at 296(2) K. The diffraction data were integrated by using the SAINT program25 and semiempirical absorption correction was applied using the SADABS program.26 The structures were solved by direct methods and refined on F2 by full-matrix least-squares methods using the SHELXTL program package.27 All non-hydrogen atoms were refined anisotropically with the hydrogen atoms added to their geometrically ideal positions and refined isotropically. The hydrogen atoms of hydroxyl groups in 1 and coordinated water molecules in 3 were located by difference Fourier map and refined by riding mode. The hydrogen atoms of lattice water molecules in 3 are split over two sites with a total occupancy of 1, which were refined by constraints using the PART command. Application of the SQEEZE routine in the PLATON software package28 produced a new intensity data set excluding the intensity contribution from disordered solvent molecules. The final chemical formula of 4 was calculated from SQUEEZE results combined with the TGA and elemental analysis data. In 1, the –(CH2)4– group was refined by using standard geometry constraint DFIX to improve the refinement stability as well as similar Uij SIMU restraint. Besides these commands, the ISOR command was utilized to restrain atom C15. Crystal data and structure refinements for 1–4 and selected bond distances and angles for 1–4 are listed in Tables 1 and S1.†
Table 1 Crystal data and structure refinements for 1–4
| R1 = ∑(|Fo| − |Fc|)/∑|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Complex |
1 |
2 |
3 |
4 |
| Molecular formula |
C22H27NiBrN4O5 |
C20H21NiBrN4O4 |
C56H54Ni4Br4N8O19 |
C24H29NiBrN5O5 |
| Fw |
566.10 |
520.03 |
1696.54 |
606.14 |
| Temperature |
296(2) |
296(2) |
296(2) |
296(2) |
| λ (Å) |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Orthorhombic |
Triclinic |
Monoclinic |
| Space group |
P21/c |
P212121 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/m |
| a (Å) |
11.1506(11) |
12.166(2) |
9.2513(9) |
8.8813(18) |
| b (Å) |
16.0135(16) |
13.298(2) |
10.0437(10) |
17.915(4) |
| c (Å) |
16.0539(12) |
13.414(2) |
17.4014(17) |
17.223(3) |
| α (°) |
90 |
90 |
76.912(2) |
90 |
| β (°) |
124.571(5) |
90 |
89.341(2) |
96.942(4) |
| γ (°) |
90 |
90 |
79.841(2) |
90 |
| V (Å3) |
2360.4(4) |
2170.1(6) |
1549.5(3) |
2720.3(9) |
| Z |
4 |
4 |
1 |
4 |
| ρ (g cm−3) |
1.593 |
1.592 |
1.819 |
1.480 |
| F(000) |
1160 |
1056 |
850 |
1244 |
| Rint |
0.0691 |
0.0659 |
0.0308 |
0.0823 |
| GOF on F2 |
1.070 |
0.941 |
1.036 |
1.057 |
| R1a[I > 2σ(I)] |
0.0629 |
0.0631 |
0.0472 |
0.0781 |
| wR2b[I > 2σ(I)] |
0.1494 |
0.1529 |
0.1117 |
0.2014 |
Results and discussion
Structure description of [Ni(bib)(5-Br-H2ip)(CH3CH2OH)]n (1)
Complex 1 crystallizes in a centrosymmetric monoclinic space group P21/c and exhibits an unusual [2 + 2] diamond construction. As described in Fig. 1a, the asymmetric unit of 1 consists of one independent Ni(II) ion, one bib ligand, one 5-Br-ip2− anion and one coordinated ethanol molecule. The Ni(II) ion is located in a distorted NiN2O4 octahedral coordination geometry and coordinated by two nitrogen atoms (N1, N4) from two different bib ligands (Ni(1)–N(1) 2.085(6), Ni(1)–N(4) 2.044(6)) and four oxygen atoms (O1, O2, O4, O5) from two different 5-Br-ip2− ligands and one ethanol (Ni(1)–O(1) 2.130(4), Ni(1)–O(2) 2.137(5), Ni(1)–O(4) 2.052(4), Ni(1)–O(5) 2.129(5)). In addition, two fully deprotonated carboxylic groups of 5-Br-ip2− adopt μ1-η1:η1 and μ1-η0:η1 coordination modes (Scheme 2a) in complex 1, linking adjacent Ni(II) ions into 1D zigzag chains with a Ni⋯Ni distance of 10.1889(14) Å and Ni⋯Ni⋯Ni angle of 132.826(12)°. The bib displays a μ2-bridging mode connecting the Ni(II) ions to also build a zigzag chain with a Ni⋯Ni distance of 14.3844(16) Å, and the dihedral angle between the two imidazole rings is 16.65° (Scheme 1a, Table 2). Using a topological approach to gain a better insight into this framework, complex 1 is a dia net topological type29 with the point symbol 66 and the long symbol 62·62·62·62·62·62, if we consider Ni(II) ions as 4-connected nodes, 5-Br-ip2− anions and bib ligands as linkages. A single adamantanoid framework is illustrated in Fig. 1b, which possesses maximum dimensions (the longest intracage distances between Ni⋯Ni) of 32.03 × 26.75 × 22.30 Å. Four independent equivalent cages to interpenetrate with each other due to the large cavity in the single net, and complex 1 exhibits an unusual [2 + 2] interpenetration mode (Fig. 1c).
 |
| | Fig. 1 (a) The coordination environment of Ni(II) ion in 1. (b) A single adamantanoid cage of 1. (c) Left: The [2 + 2] interpenetrating dia network viewed along the c axis. Right: Schematic representation of the [2 + 2] interpenetrating dia network. | |
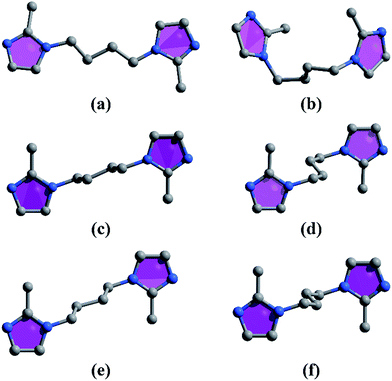 |
| | Scheme 1 The conformations of the bib ligand in complexes 1–4: (a) for 1, (b) for 2, (c) and (d) for 3, (e) and (f) for 4. | |
Table 2 Distance between two coordinated N atoms of the bib ligand and dihedral angle of two imidazole rings
| Complex |
Distance between two coordinated N atoms (Å) |
Dihedral angle of two imidazole rings (°) |
| 1 |
10.43 |
16.65 |
| 2 |
6.82 |
25.74 |
| 3 |
10.19 |
0 |
| 8.92 |
0 |
| 4 |
10.22 |
0 |
| 8.60 |
0 |
Structure description of [Ni(bib)(5-Br-ip)]n (2)
Complex 2 crystallizes in chiral orthorhombic space group P212121 and shows a 3D non-interpenetrated structure. As shown in Fig. 2a, the asymmetric unit of 2 consists of one Ni(II) ion, one fully deprotonated 5-Br-ip2− anion and one bib ligand. The Ni(II) ion is located in a distorted octahedral coordination geometry and coordinated by two nitrogen atoms (N1, N4) from two bib ligands (Ni(1)–N(1) 2.002(6), Ni(1)–N(4) 2.019(7)) as well as four oxygen atoms (O1, O2, O3, O4) from two 5-Br-ip2− ligands (Ni(1)–O(1) 2.090(5), Ni(1)–O(2) 2.189(6), Ni(1)–O(3) 2.100(6), Ni(1)–O(4) 2.154(6)). Each bib acts as a bridging ligand (with the dihedral angle of 25.74° between the two imidazole rings) (Scheme 1b, Table 2) coordinating to Ni(II) centers to form a zigzag chain with the Ni⋯Ni separation of 8.1252(21) Å and Ni⋯Ni⋯Ni angle of 96.975(21)° (Fig. 2b). The two carboxylic groups of 5-Br-ip2− display the same μ1-η1:η1 coordination mode (Scheme 2b) to bridge the Ni(II) centers to form a 1D left-handed {[Ni(5-Br-ip)2−]}n helical chain along the b axis with a helical pitch of 13.2540(26) Å (Fig. 2c). Furthermore, these zigzag chains and the 1D left-handed helical chains are linked by each other to generate a 2D net (Fig. 2d) and further connected along the c axis to give a 3D framework. From the topological point of view, each six-coordinated Ni(II) ion in complex 2 can be considered as a 4-connected node to link three neighboring Ni(II) ions together, and thus complex 2 also exhibits a dia topology29 with the point symbol 66 and the long symbol 62·62·62·62·62·62 (Fig. 2e).
 |
| | Fig. 2 (a) The coordination environment of Ni(II) ion in 2. (b) Perspective view of zigzag chain in 2. (c) Perspective view of left-handed helical chain in 2. (d) A 2D net (e) The dia network. | |
 |
| | Scheme 2 The coordination modes of the 5-Br-ip2− ligand in complexes 1–4: (a) for 1, (b) for 2 and 4, (c) and (d) for 3. | |
Structure description of {[Ni2(bib)(5-Br-H2ip)2H2O]·0.5H2O}n (3)
Complex 3 crystallizes in the centrosymmetric triclinic space group P and features a dinuclear 3D pillar-layer network in which the Ni⋯Ni separation is 3.4347(11) Å. The asymmetric unit of 3 is composed of two Ni(II) ions, one bib ligand, two 5-Br-ip2− anions, one coordinated water molecule and a half lattice water molecule (Fig. 3a). Ni1 is bonded to five oxygen atoms (O1, O3, O4, O5, O7) from four 5-Br-ip2− ligands (Ni(1)–O(1) 2.010(3), Ni(1)–O(3) 2.100(3), Ni(1)–O(4) 2.111(3), Ni(1)–O(5) 2.081(3), Ni(1)–O(7) 2.106(3)) and one nitrogen atom (N1) from a bib ligand (Ni(1)–N(1) 2.098(4)) to finish a distorted octahedral geometry. Ni2 also with distorted octahedral geometry is six-coordinated by five oxygen atoms (O2, O6, O7, O8, O1W) from three 5-Br-ip2− ligands (Ni(2)–O(2) 2.014(3), Ni(2)–O(6) 2.018(3), Ni(2)–O(7) 2.045(3), Ni(2)–O(8) 2.380(4), Ni(2)–O(1W) 2.127(4)) and one water molecule, and one nitrogen atom (N4) from a bib ligand (Ni(2)–N(4) 2.050(4)). The 5-Br-ip2− in 3 displays two kinds of coordination modes; one 5-Br-ip2− connects to three Ni(II) ions through two carboxylic groups which adopt μ2-η1:η1 and μ1-η1:η1 coordination modes (Scheme 2c), while the other links to four Ni(II) ions in which the carboxylic groups adopt μ2-η1:η1 and μ2-η1:η2 coordination modes (Scheme 2d), and that to induce a (4,4) grid sheet (Fig. 3b). Then the 2D layers are further connected by bib to yield a 3D pillar-layer framework (Fig. 3c). It is interesting that the bib adopts two different trans conformations, due to flexible –(CH2)4– groups (Scheme 1c and d, Table 2). According to the simplification principle, the dimeric units can be regarded as six-connected nodes, thus the resulting structure of 3 is a six-connected pcu net29 with a point symbol of 412·63 (Fig. 3d).
 |
| | Fig. 3 (a) The coordination environment of Ni(II) ion in 3. (b) (4,4) grid sheet. (c) The 3D pillar-layer framework. (d) The pcu network. | |
Structure description of {[Ni(bib)(5-Br-H2ip)]·DMA}n (4)
Complex 4 crystallizes in a centrosymmetric monoclinic space group P21/m and displays a 2D stacked layer. The asymmetric unit of 4 contains one Ni(II) ion, one 5-Br-ip2− anion, one bib ligand and one guest DMA molecule (Fig. 4a). Each Ni(II) ion is six-coordinated by four oxygen atoms (O1, O2, O3, O4) from two 5-Br-ip2− ligands (Ni(1)–O(1) 2.060(5), Ni(1)–O(2) 2.248(5), Ni(1)–O(3) 2.243(5), Ni(1)–O(4) 2.058(5)) and two nitrogen atom (N1, N4) from two bib ligands (Ni(1)–N(1) 2.040(6), Ni(1)–N(4) 2.034(6)), forming a distorted octahedral coordination geometry. The 5-Br-ip2− ligand employs the μ1-η1:η1 coordination mode (Scheme 2b) to join two Ni(II) ions to yield an infinite 1D linear chain with a Cd⋯Cd⋯Cd angle of 180.00°. Such 1D chains are linked by bib (Scheme 1e and f, Table 2), giving rise to a 2D wave-like layer. And the 2D layers further connect with each other via π–π interactions between the adjacent imidazole rings of bib ligands with d = 3.36 Å, θ = 0° (d is on behalf of the centroid–centroid distance, and θ is the dihedral angle between two imidazole rings) (Fig. 4b) and stack in an AAA fashion resulting into a 3D supramolecular framework (Fig. 4c), which possesses the 1D channel with the guest DMA molecules. After the guest DMA molecules were omitted, solvent-accessible volume of 4 is 30.3%, as determined from the PLATON program.28 Considering Ni(II) ions as 4-connected nodes, while both bib and 5-Br-ip2− serve as linkers, the whole structure of 4 can be defined as a sql net29 with the point symbol 44·62 (Fig. 4d).
 |
| | Fig. 4 (a) The coordination environment of Ni(II) ion in 4. (b) The π–π stacking interactions. (c) The 3D supramolecular framework. (d) The sql network. | |
Influence of solvent in assembly process and comparison of structures
From the structural description above, we found that the solvent has a crucial effect on the final structure of CPs. For 1, EtOH molecules as terminal ligand impact on the network structure. The EtOH molecule occupies the axial position of the octahedral Ni(II) center, which lead to two carboxylic groups of 5-Br-ip2− respectively adopt μ1-η1:η1 and μ1-η0:η1 coordination modes link two Ni(II) ions. Complexes 2 and 3 were synthesized under similar conditions by using different solvent systems MeCN–EtOH and MeCN–H2O. The structural discrepancies between 2 and 3 may be attributed to polarity and size of the solvent molecules. In 2, the solvent molecule absent in the final structure, whereas H2O molecules are coordinated to Ni(II) ions in 3. The results correspond to the polarity and coordinating ability of the solvents H2O > EtOH.30 Compared with EtOH (31.94 cm3 mol−1), H2O has a smaller van der Waals volume31 (11.44 cm3 mol−1), which makes molecules collide more effectively, thus resulting in the more complicated dinuclear 3D network. For 4, DMA as guest molecules induces the formation of the 2D layer framework. The steric hindrance of guest DMA molecules makes it difficult to further extend to a 3D framework and induces a high solvent-accessible volume. It is apparent that the CPs assemblies were extremely sensitive to solvent systems.
PXRD and TGA
The X-ray powder diffraction patterns show that the diffraction peaks of both the simulated and experimental patterns match well in relevant positions, indicating the phase purities of these complexes (Fig. S1–S4†). Thermogravimetric experiments were conducted to examine the thermal stabilities of complexes 1–4 (Fig. S5–S8†). The TGA curve of 1 indicates that a weight loss of 7.64% appears before 230 °C corresponds to the release of coordinated ethanol molecules (calcd 8.13%), and the decomposition of the residual framework occurs at 280 °C. For 2, the framework remains stable to about 290 °C. After that temperature, the residue starts to collapse. For 3, the weight loss of 2.90% (calcd 3.18%) below 325 °C corresponds to the loss of coordinated water molecules and lattice water molecules. For 4, the curve displays a decrease of 14.36% before 215 °C, suggesting the departure of the corresponding guest DMA molecules (calcd 14.37%). Then the framework begins to collapse at 305 °C.
Magnetic property
The variable-temperature magnetic susceptibility of complexes 1–4 were measured at a field of 1000 Oe in the temperature range of 2–300 K. The magnetic susceptibilities χM and χMT vs. T plots are shown in Fig. 5. For 1 and 4, the experimental χMT values are 1.67 cm3 mol−1 K and 1.26 cm3 mol−1 K at 300 K, respectively, which are larger than the spin only value Ni(II) ion (1.0 cm3 mol−1 K, S = 1, g = 2.0) due to the orbital contribution to the magnetic moment.32 As the temperature is lowered from 300 K, the χMT values decrease smoothly to 1.47 cm3 mol−1 K at 16 K and 1.15 cm3 mol−1 K at 18 K, then decreasing abruptly to the values of 0.90 cm3 mol−1 K and 0.77 cm3 mol−1 K at 2.0 K. The temperature dependence of the reciprocal susceptibilities (1/χM) obeys the Curie–Weiss law χM = C/(T − θ) with C = 1.62 cm3 mol−1 K, θ = −3.83 K for 1 and C = 1.26 cm3 mol−1 K, θ = −2.71 K for 4. The behavior of the χMT values and the negative θ values indicate the presence of antiferromagnetic interaction between the Ni(II) ions.
 |
| | Fig. 5 Temperature dependence of χMT and χM vs. T plots of 1 (a), 2 (b), 3 (c) and 4 (d). Inset: temperature dependence of χM−1; the solid line represents the best fit of the Curie–Weiss law. | |
For 2, at 300 K, the experimental χMT value is equal to 1.34 cm3 mol−1 K, which is slightly higher than the spin only value Ni(II) ion (1.0 cm3 mol−1 K, S = 1, g = 2.0).33 Up cooling, the χMT value remains almost constant until 25 K, revealing a typical paramagnetic behavior.34 Upon further cooling, a sharp decrease is observed, and the χMT value reaches a minimum of 0.67 cm3 mol−1 K at 2 K, which suggests the antiferromagnetic interactions in 2. The magnetic data between 2 to 300 K were fitted with the Curie–Weiss law χM = C/(T − θ) yielding the values of C = 1.34 cm3 mol−1 K, θ = −1.60 K. The negative θ value confirms that there is an antiferromagnetic interaction between the Ni(II) ions.
For 3, the value of χMT at 300 K is 3.38 cm3 mol−1 K, is higher than the spin only value of 2.0 cm3 mol−1 K expected for two magnetically isolated high-spin Ni(II) ions (S = 1, g = 2.0).32 Upon lowering the temperature, the value of χMT decreases gradually to 3.14 cm3 mol−1 K at 20 K, and then at lower temperatures decreases more steeply to reach a value of 1.29 cm3 mol−1 K at 2.0 K, indicating an antiferromagnetic coupling between Ni2 dimers. The Curie's–Weiss fit in the range of 2.0 to 300 K obtained the values of C = 3.37 cm3 mol−1 K and θ = −2.54 K. The results of negative θ value indicate that there is an antiferromagnetic exchange between the neighboring Ni centers.
Conclusions
In conclusion, four new solvent-controlled isomeric Ni(II) CPs were successfully synthesized under solvothermal conditions. In the self-assembly process of the four CPs, the solvent media play a diverse role, which influences coordination number and geometry of metal centers, conformation of the flexible bib ligands and coordination mode of 5-Br-ip2− ligands, resulting in different structures. This study provides us a possible approach to designing and fabricating novel CPs.
Acknowledgements
We are grateful for financial support from the NSF of China (No. 21373122), the NSF for Fostering Talents in Basic Science (No. J1210057), the NSF of Shaanxi/Hubei Province (Grant 11JK0590 and Z2011CDA118), and the Open Foundation of Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education (338080057).
Notes and references
- S. Wang, T. T. Zhao, G. H. Li, L. Wojtas, Q. S. Huo, M. Eddaoudi and Y. L. Liu, J. Am. Chem. Soc., 2010, 132, 18038–18041 CrossRef CAS PubMed
 .
. - X. F. Kuang, X. Y. Wu, R. M. Yu, J. P. Donahue, J. S. Huang and C. Z. Lu, Nat. Chem., 2010, 2, 461–465 CrossRef CAS PubMed .
- D. S. Li, Y. P. Wu, J. Zhao, J. Zhang and J. Y. Lu, Coord. Chem. Rev., 2014, 261, 1–27 CrossRef CAS PubMed .
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC .
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC .
- L. N. Li, J. X. Ma, C. Song, T. L. Chen, Z. H. Sun, S. Y. Wang, J. H. Luo and M. C. Hong, Inorg. Chem., 2012, 51, 2438–2442 CrossRef CAS PubMed .
- B. Liu, W. P. Wu, L. Hou and Y. Y. Wang, Chem. Commun., 2014, 50, 8731–8734 RSC .
- L. J. Li, S. F. Tang, C. Wang, X. X. Lv, M. Jiang, H. Z. Wu and X. B. Zhao, Chem. Commun., 2014, 50, 2304–2307 RSC .
- C. P. Li, J. M. Wu and M. Du, Chem.–Eur. J., 2012, 18, 12437–12445 CrossRef CAS PubMed .
- S. B. Li, H. Y. Ma, H. J. Pang and L. Zhang, Cryst. Growth Des., 2014, 14, 4450–4460 CAS .
- L. Y. Du, W. J. Shi, L. Hou, Y. Y. Wang, Q. Z. Shi and Z. H. Zhu, Inorg. Chem., 2013, 52, 14018–14027 CrossRef CAS PubMed .
- S. J. Yang, J. H. Cho, K. Lee, T. Kim and C. R. Park, Chem. Mater., 2010, 22, 6138–6145 CrossRef CAS .
- Y. W. Li, D. C. Li, J. Xu, H. G. Hao, S. N. Wang, J. M. Dou, T. L. Hu and X. H. Bu, Dalton Trans., 2014, 43, 15708–15712 RSC .
- W. W. Dong, D. S. Li, J. Zhao, L. F. Ma, Y. P. Wu and Y. P. Duan, CrystEngComm, 2013, 15, 5412–5416 RSC .
- X. R. Hao, X. L. Wang, K. Z. Shao, G. S. Yang, Z. M. Su and G. Yuan, CrystEngComm, 2012, 14, 5596–5603 RSC .
- C. P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC .
- X. Y. Liu, P. P. Cen, H. Li, H. S. Ke, S. Zhang, Q. Wei, G. Xie, S. P. Chen and S. L. Gao, Inorg. Chem., 2014, 53, 8088–8097 CrossRef CAS PubMed .
- T. Wang, C. L. Zhang, Z. M. Ju and H. G. Zheng, Dalton Trans., 2015, 44, 6926–6935 RSC .
- S. C. Chen, Z. H. Zhang, K. L. Huang, Q. Chen, M. Y. He, A. J. Cui, C. Li, Q. Liu and M. Du, Cryst. Growth Des., 2008, 8, 3437–3445 CAS .
- J. Li, G. P. Yang, L. Hou, L. Cui, Y. P. Li, Y. Y. Wang and Q. Z. Shi, Dalton Trans., 2013, 42, 13590–13598 RSC .
- Y. F. Hou, B. Liu, K. F. Yue, C. S. Zhou, Y. M. Wang, N. Yan and Y. Y. Wang, CrystEngComm, 2014, 16, 9560–9567 RSC .
- M. L. Han, J. G. Wang, L. F. Ma, H. Guo and L. Y. Wang, CrystEngComm, 2012, 14, 2691–2701 RSC .
- X. J. Li, Z. J. Yu, T. N. Guan, X. X. Li, G. C. Ma and X. F. Guo, Cryst. Growth Des., 2014, 15, 278–290 Search PubMed .
- M. Arıcı, O. Z. Yeşilel and M. Taş, Cryst. Growth Des., 2015, 15, 3024–3031 Search PubMed .
- SAINT, Program for Data Extraction and Reduction, Bruker AXS, Inc., Madison, WI, 2001 Search PubMed .
- G. M. Sheldrick, SADABS, University of Göttingen, Göttingen, Germany Search PubMed .
- G. M. Sheldrick, SHELXTL, Version 6.10, Bruker Analytical X-ray Systems, Madison, WI, 2001 Search PubMed .
- A. L. J. Spek, Appl. Crystallogr., 2003, 36, 7 CrossRef CAS .
- V. A. Blatov, Struct. Chem., 2012, 23, 955 CrossRef CAS .
- R. Diaz-Torres and S. Alvarez, Dalton Trans., 2011, 40, 10742–10750 RSC .
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS .
- W. H. Yan, S. S. Bao, J. Huang, M. Ren, X. L. Sheng, Z. S. Cai, C. S. Lu, Q. J. Meng and L. M. Zheng, Dalton Trans., 2013, 42, 8241–8248 RSC .
- S. H. Zhang, N. Li, C. M. Ge, C. Feng and L. F. Ma, Dalton Trans., 2011, 40, 3000–3007 RSC .
- X. H. Chen, Q. J. Wu, W. Lu, M. X. Yang and L. J. Chen, Inorg. Chem. Commun., 2011, 14, 694–696 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns and TGA. CCDC 1410660–1410663. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra15397b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 






