
Photocurrent generation of nanofibers constructed using a complex of a gelator and a fullerene derivative†
Abstract
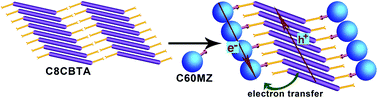
A carboxylic acid derivative (C8CBTA) with benzothiadiazole as an electron-withdrawing unit and N-octyl 3-amidocarbazole as an electron donor was designed and synthesized to obtain a gelator with a longer absorption wavelength. C8CBTA could form red gels in some aromatic solvents and could self-assemble into thin nanofibers in gel phases. The changes in UV-vis absorption spectra during gelation suggested J-aggregate stacking between aromatic moieties, and the IR spectrum of the xerogel film revealed the intermolecular hydrogen bonding between amide groups and the formation of dimers between carboxyl groups in gel phases. Thus, the synergistic effect of π–π interaction and hydrogen bonding induced 1D stacking. The C8CBTA gel exhibited a maximal absorption peak at 520 nm and emitted weak near-infrared fluorescence at a maximum of 700 nm because of the strong intramolecular charge transfer. A fullerene derivative that contains an imidazole unit as a hydrogen bond acceptor was synthesized and used as an electron acceptor in constructing a photo-induced electron-transfer two-component gel with C8CBTA. Fullerene derivatives also formed 1D stacking in the two-component gel. Moreover, the xerogel film, which served as the active layer, generated a larger amount of photocurrent compared with the disordered film under light irradiation.
 Please wait while we load your content...
Please wait while we load your content...

