
Enzyme activity enhancement of chondroitinase ABC I from Proteus vulgaris by site-directed mutagenesis†
Abstract
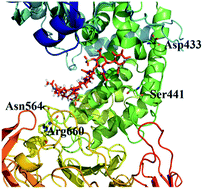
Chondroitin sulfate (CS) is widely applied in the medical industry, especially CS B which is a kind of CS and widely used in the field of food industry, medicine and scientific research. Because of the high molecular weight of CSs, many functions could not be realized effectively. Chondroitinase ABC I (ChSase ABC I) could degrade CS to low molecular weight CS. In this study, ChSase ABC I was expressed with a maltose binding protein (MBP) tag, and site-directed mutagenesis based on both sequence alignment and molecular docking simulation analysis was conducted. 13 amino acids were selected to be mutated to Ala separately for the first time, 8 out of the 13 single-amino-acid mutants showed decreased activity with CS A as substrate and 11 of them showed decreased activity with CS B as substrate. Mutating Arg660 to Ala caused a total loss of the enzyme activity either with CS A or CS B as substrate, indicating that Arg660 was one of the active sites of ChSase ABC I. The specific activities of Asn795Ala and Trp818Ala were 1.39 and 1.38 times higher than that of wild type enzyme with CS A as substrate, and 1.85 and 1.71 times higher with CS B as substrate. In particular, the specific activity of Asn795Ala in this study was the highest among the reported ones. The kinetic parameters as well as the thermostabilities of the two mutants also showed significant improvement when compared with those of the wild type enzyme.
 Please wait while we load your content...
Please wait while we load your content...

