
Recent progress towards ionic hydrogenation: Lewis acid catalyzed hydrogenation using organosilanes as donors of hydride ions
Tianqi Liuab,
Xiaojian Wang*ab and
Dali Yin*ab
aState Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing 100050, P.R. China. E-mail: wangxiaojian@imm.ac.cn; Fax: +86 10 63165248; Tel: +86 10 63165248
bDepartment of Medicinal Chemistry, Beijing Key Laboratory of Active Substances, Discovery and Drugability Evaluation, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing 100050, P.R. China
First published on 1st September 2015
Abstract
Development of the methodology for hydrogenation is of great significance in organic chemistry. Ionic hydrogenation is attractive for its chemoselectivity and unique feature of product structures which is a result of the combination of reduction, cyclization and intermolecular addition. It is mostly suitable for the reduction of carbonyl groups, unsaturated hydrocarbons, imines and reductive amination of carbonyl compounds. Recent advances in ionic hydrogenation as well as its fundamental mechanism are summarized and discussed.
Introduction
Hydrogenation is one of the fundamental reactions in modern organic synthesis, which has experienced impressive progress during the last few decades.1–3 From a practical perspective, hydrogenations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN and CO bonds are used widely, and are particularly prevalent in the synthesis of fine chemicals as well as compounds used in pharmaceutical and agricultural industries.4–6
C, CN and CO bonds are used widely, and are particularly prevalent in the synthesis of fine chemicals as well as compounds used in pharmaceutical and agricultural industries.4–6
Hydrogenation mainly falls into three types, including catalytic hydrogenation, metal hydrides reduction and Lewis catalysed ionic hydrogenation. Traditionally, catalytic hydrogenation and metal hydride reduction are the most prevalent methods used. The chemoselectivity of each method varies depending on different catalysts selection or on modification of hydrides. By reason of the environmental friendliness and low cost of H2, homogeneous and heterogeneous catalytic reactions using molecular hydrogen are attractive in organic chemistry. Catalytic hydrogenation tends to reduce less polar double or triple bonds, such as CC, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, CN, NO2, etc.7,8 The heterogeneous hydrogenation is carried out efficiently on supported metals since solid catalysts are easy to separate and recycle.9 Many catalysts have been developed, such as RANEY®-Ni, Zn–Hg/HCl, Lindlar catalyst, etc.10–13 Nevertheless, the activity and chemoselectivity vary on the catalysts as well as the reaction condition.14
C, CN, NO2, etc.7,8 The heterogeneous hydrogenation is carried out efficiently on supported metals since solid catalysts are easy to separate and recycle.9 Many catalysts have been developed, such as RANEY®-Ni, Zn–Hg/HCl, Lindlar catalyst, etc.10–13 Nevertheless, the activity and chemoselectivity vary on the catalysts as well as the reaction condition.14
Meanwhile, metal hydrides reagents favour more polar groups like COOR, and CO etc.15,16 Among which, LiAlH4 and NaBH4 are often used in the reduction of ketones to alcohols.17,18 However, they consume stoichiometric amounts of the hydride reagent and produce waste by-products. Therefore, these types of reactions would not be environmentally friendly synthetic processes. In addition, a large percentage of the known methods demand the presence of transition metals as catalysts. RuII complexes have been shown to be effective in hydrogenation of CC bonds of maleic acid and nitriles,19,20 but the classic mechanism mainly relies on the ability of the metal hydride bond to insert unsaturated bonds, which leads to the limitation of catalytic pathways in hydrogenation. Hence, although significant progress has been made in hydrogenation, there is still a great need for new methods that can overcome such problems.
Intense study of hydrogenation reactions continues, where a special place among methods of hydrogenation is occupied by ionic hydrogenation. It was first reported in 1974 by Kursanov and coworkers, who pioneered the use of CF3COOH as the H+ source and Et3SiH as the hydride donor in the hydrogenation of ketone and other unsaturated organic compounds.21 After almost 30 years of dreariness, ionic hydrogenation has received considerable attention with impressive advancement in recent years due to its unique features in reduction conditions, substrate scope, and special product structures. It refers to the hydrogenation implemented by the addition of a proton and a hydride ion to a substrate, permitting the hydrogenation of alkenes, ketones, aldehydes, anils and imines. Various Lewis acids and hydride ion donors can be employed in the ionic hydrogenation. With the proper selection of a hydrogenating pair or catalyst, high reactivity is generally achieved under mild reaction conditions with broad range of substrate scope and excellent functional group tolerance.22,23 More importantly, some special products can be obtained by using ionic hydrogenation, including diverse heterocycle building blocks. Regarding the reaction mechanism, the addition of a proton to generate a cation is the first step of this procedure, which then reacts with a hydride ion to give the corresponding hydrogenation product.22 When adopting the organosilane and Lewis acid as the hydrogenating pair, the mechanism can be described as follows (Fig. 1):24

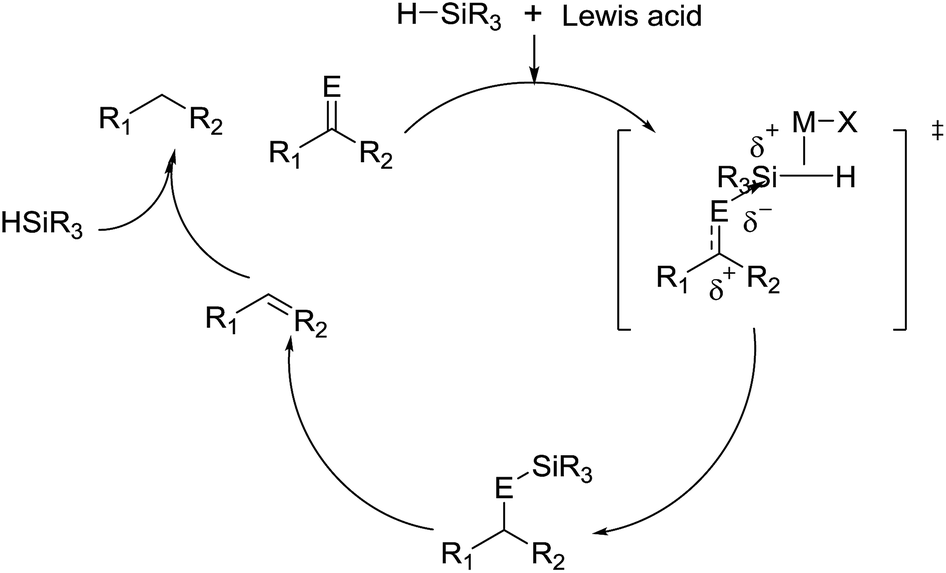 | ||
| Fig. 1 General reaction mechanism. | ||
In this review, we will focus on recent advances in Lewis acids catalyzed ionic hydrogenation using organosilanes as donor of hydride ion, especially for its utilization in the reduction of carbonyl group, imine, unsaturated hydrocarbon and halogenated hydrocarbon since 1995, as well as the discussion of specific reaction mechanisms. Selected examples of substrates will be listed likewise, and a personal outlook will be given at the end.
Reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O bond
O bond
Each of the proposed methods of hydrogenation has fundamental limitations as well as its own merits. Compared with other traditional reduction approaches, ionic hydrogenation is a powerful tool for the hydrogenation of carbonyl compounds for its high chemoselectivity and wide availability, which can be carried out by silane and Lewis acid.
In 2011, Norio Sakai and coworkers reported an InBr3–Et3SiH mediated hydrogenation of aliphatic ketones to give secondary alcohols selectively in moderate to good yields (Scheme 1).25 Among the catalysts authors examined, only InBr3 showed outstanding catalytic activity, while InCl3 and In(OAc)3 were inefficient for the reduction. It's noteworthy that although the hydroxyl group on the benzene ring was silylated during the reduction of ketones, silyl could be easily removed to afford the theoretical products. This reaction exhibited a good functional group tolerance, for double bond and cyano group were all compatible with the reducing system. Interestingly, symmetrical ether derivatives could be produced in good yields by changing the order of addition of the reagents, in which the initial mixing of InBr3 and PhSiH3 (or Et3SiH) was followed by the addition of an aliphatic ketone and solvent. Additionally, the reducing system was tested for the hydrogenation of aromatic ketone and acetophenone derivatives, deoxygenation proceeded preferentially to give corresponding alkyl benzene derivatives as well (Scheme 2). The formation of products was influenced by many factors like electron-withdraw group and bromomethylene moiety, where the corresponding silyl ether product was obtained rather than the deoxygenative product.
 | ||
| Scheme 1 Selective conversion from aliphatic ketones to secondary alcohols and symmetrical ethers. | ||
 | ||
| Scheme 2 Selective reduction of aromatic ketones. | ||
To further proving the efficiency of ionic hydrogenation in aromatic ketone, in 2014, Xiao-jun Wang and coworkers fulfilled the facial reduction of benzophenones (8) using aluminum chloride (AlCl3) in combination with 1,1,3,3-tetramethyldisiloxane in the production of SGLT-2 inhibitor Empagliflozin (Scheme 3).26 They extended the result to the reduction of pyranoside to selectively afford β-C-glycosides. With the addition of methyl β-glycopyranoside (4) to Et3SiH and AlCl3, acceptable results were produced compared to BF3·OEt2 (Scheme 4). What's more, the reaction using AlCl3 as a Lewis acid was much less sensitive to water content, conductive to avoid side reaction.
 | ||
| Scheme 3 Reduction of benzophenones. | ||
 | ||
| Scheme 4 Reduction of methyl β-glycopyranoside. | ||
In the same year, Tomoko Mineno and coworkers reported the utility of Et3SiH and In(OTf)3 in the reductive etherification of aldehydes, similar to Norio Sakai's research mentioned above (Scheme 5).27 Under the optimized condition comprised of Et3SiH and In(OTf)3 using CH2Cl2 as the solvent, a wide range of substrates such as benzaldehyde derivatives, bicyclic aromatics, aliphatic aldehydes can undergo this reaction to construct symmetrical ethers in good to excellent yields. Besides, substrates bearing protecting group of hydroxyl succeeded in undergoing the reductive coupling reaction, giving corresponding ethers without cleavage of the protection.
 | ||
| Scheme 5 Reductive coupling reaction of aldehydes. | ||
Not only simple carbonyl compounds can be hydrogenated to give alkanes, alcohols, and ethers, a wide array of esters and amides can also undergo the reduction successfully. In 2007, Norio Sakai and coworkers developed an InBr3/Et3SiH catalyzed direct reductive deoxygenation of esters for the synthesis of unsymmetrical ethers (Scheme 6).28 The reagent system exhibited excellent functional group tolerance, including the introduction of nitro and halogen group. In addition, a broad range of carboxylic acid, aromatic, aliphatic, heteroaromatic and cyclic esters can produce ethers with good yields. If reversing the location of ester group, the reaction was also carried out smoothly. Notably, the acetoxy group proceeded deacetoxylation preferentially when bonding to a phenyl group or a conjugated double bond. A plausible radical-based mechanism was proposed for the reductive deoxygenation of esters.
 | ||
| Scheme 6 Reductive deoxygenation of esters. | ||
Later on, the same group described a reductive deoxygenation of tertiary amides (Scheme 7).29 With the reaction using 5% InBr3 and 4 equiv. of Et3SiH as the reagent system, a variety of tertiary amines were constructed. When changing the Lewis acid or hydrosilane, decrease of catalytic activity and product yield occurred apparently. A wide scope of substrates such as benzamide, acetanilide, aliphatic, heteroaromatic amide derivatives can undergo this reaction in excellent yields. Additionally, this approach can also be applied to the reduction of the carbonyl of secondary amides. In the proposed mechanistic path of the reaction, the amide first reacts with HInBr2 to form an N,O-acetal intermediate, which further transforms to the imine in the presence of InBr3, subsequent reduction of the iminium ion provides the corresponding amine (Fig. 2).
 | ||
| Scheme 7 Reductive deoxygenation of tertiary amides and partial secondary amides. | ||
 | ||
| Fig. 2 Proposed reaction mechanism. | ||
As mentioned before, apart from tertiary amides, ionic hydrogenation is also suitable for secondary amides. In 2012, an iridium-catalyzed reduction of secondary amides using diethylsilane was reported by Chen Cheng's group, which accomplished a further research on the reduction deoxygenation of amides (Scheme 8).30 A broader range of substrates with diverse functionalities has been reduced to secondary amines and imines in high yields. However, excluding nitro and nitrile groups, where little target products can be obtained or the conversion to amines is low. Based on the study of catalyst resting state, the authors proposed the reaction beginning with the hydrosilylation of the CO bond, followed by elimination of silanol to give the imine. Consecutive hydrosilylation of the imine CN bond occurs, finally leading to the corresponding amine after the acidification of aqueous HCl workup.
 | ||
| Scheme 8 Reduction of secondary amides. | ||
Other than the direct reduction of various carboxides via ionic hydrogenation, TFA/Et3SiH-mediated dehydrative reduction plays an important role in the cyclization of thioketones, producing syn-bisaryl (or heteroaryl)dihydrobenzoxathiins and benzodioxane with high diastereoselectivity, in good yields. Seongkon Kim and coworkers also examined BF3 etherate as the reagent system, but the former achieved better yields (Scheme 9).31 By employing 1H NMR, the cis–trans configuration of the product was determined, which proved the total diastereoselectivity (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Interestingly, the yield and reaction time was sensitive to the steric and electronic environment. For a substrate with meta-benzyloxy, lower yields obtained and longer reaction times required, similar to fluoro-substituted thioketones. In addition, they found that the procedure can also be applied to the cyclization of electron-rich heteroaromatics with good yields.
:1). Interestingly, the yield and reaction time was sensitive to the steric and electronic environment. For a substrate with meta-benzyloxy, lower yields obtained and longer reaction times required, similar to fluoro-substituted thioketones. In addition, they found that the procedure can also be applied to the cyclization of electron-rich heteroaromatics with good yields.
 | ||
| Scheme 9 Dehydrative reduction of thioketones. | ||
Later in 2004, Tarek Sammakia and coworkers developed a highly selectively reductive cyclization of acyclic β-alkoxy ketones using Et3SiH and SnCl4 as the reagent system to yield protected syn-1,3-diols via an oxocarbenium intermediate, which consequently broadened the application of ionic hydrogenation method (Scheme 10).32 It was found that the electronic and steric properties of the substituents had little effect on the yield and diastereoselectivity of the reaction. Various ketones can undergo the reaction in moderate to good yield and afforded >200:1 selectivity.
 | ||
| Scheme 10 Selectively reduction of acyclic β-alkoxy ketones. | ||
Interestingly, the usage of ionic hydrogenation has also been extended into the Teralins and heterocyles preparation. In these reaction processes, the hydrogenation and condensation could be achieved in one pot, which efficiently give the corresponding products in high yield. In 2011, a facile reductive cyclization of phenylpentane-1,4-diones using TiCl4/Et3SiH as catalyst was reported by Wang and coworkers (Scheme 11).33 Unlike its traditional utility in reduction area, this methodology provided a new approach for the synthesis of tetrahydronaphthalene through ionic hydrogenation and titanium(IV) chloride catalyzed cyclization. In addition, for substrates in different electronic environment, the molecules with electron-donating groups gave higher yields, in contrast to electron insufficient molecules with halogen substituents. For the mechanistic insights, they proposed that the reduction of aliphatic carbonyl occurred prior to the benzyl carbonyl to form siloxane, followed by the cyclization of siloxane to generate the 1-methyl-tetrahydronaphthalene.
 | ||
| Scheme 11 Reductive cyclization of phenylpentane-1,4-diones. | ||
Based on the progress above, they intended to apply this approach to the synthesis of other bioactive heterocycles, such as fused bicyclic oxygen-containing heterocycles, etc. In 2015, the report from Yin's group showed that this transformation can be achieved through tandem ionic hydrogenation, ketalization, and intramolecular cyclization in mild conditions (Scheme 12).34 The corresponding tetrahydrofuro[3,2-d]oxazole were stereoselectively obtained in moderate to good yields by using TiCl4/Et3SiH as the catalyst. Besides, a wide range of arene-1,4-diones bearing various functional groups on the aryl ring are compatible to this novel protocol, where electron-donating groups are more favorable. For the reaction pathway, the author proposed that the cascade process was started by the reduction of benzyl carbonyl, afterwards the ketalization and cyclization are carried out to give the carbocation intermediate, which can be further converted to desired product promoted by the triethylsilanolate anion (Fig. 3).
 | ||
| Scheme 12 Reductive cyclization of arene-1,4-diones. | ||
 | ||
| Fig. 3 Proposed reaction mechanism. | ||
Reduction of CN bond
Amines constitute an important class of chemical compounds which are widely used in industry, pharmaceutics and agrochemistry. Their direct formation via reduction of CN bond is an important transformation in academic research, and has become a useful synthetic tool of amines in organic chemistry. Recent studies have shown that ionic hydrogenation can not only achieve the reduction of carbonyl, but can also be used in the hydrogenation of imine and reductive amination of carbonyl compounds.
In 2005, Ana C. Fernandes and coworkers reported a novel method for the reduction of imines by use of the catalytic system silane/MoO2Cl2 (Scheme 13).35 After the optimization of reaction conditions, the best result was obtained in the presence of 10 mol% of MoO2Cl2 in THF at reflux temperature, with polymethylhydrosiloxane (PMHS) as the reductant. Various imines were reduced in excellent to moderate yields and chemoselectivity, especially for the substrates bearing electron-withdrawing groups, such as fluoro, chloro, ester or nitro group.
 | ||
| Scheme 13 Reduction of imines to amines. | ||
Hydrosilylation of imines also plays an important role in sequential catalysis to generate secondary bulky amines. With the employment of a hydrogenating pair or catalyst, simple imines can be transformed into amines smoothly. Many transition metal complexes have been used as catalysts for imine hydrosilylation, including Ru, Rh, Ti, Ir and Sn. The hydrosilylation of imines with Ph2SiH2 proceeded efficiently using [RuCl2(arene)]2 catalyst at room temperature to give secondary amines in moderate yields.36
Despite the importance of amines in pharmaceutical industries, they are promising raw materials and intermediates of chemical synthesis as well. During the past decade, a great deal of efforts have been devoted to the construction of carbon–nitrogen bond, among which intermolecular and intramolecular reductive amination is an indispensable method.
In 2001, Richard Apodaca and coworkers described a dibutyltin dichloride-catalyzed direct reductive amination of aldehydes and ketones using phenylsilane as reductant, succeeding in applying ionic hydrogenation in the synthesis of amines (Scheme 14).37 In the respect of the substrate scope, a wide range of carbonyl compounds with reducible functional groups or potentially acid-labile groups were well tolerated, the desired tertiary amines were produced selectively while no other reduction products were detected. Although the steric hindrance and electronic effect decreased the rate of reduction, good yields of amines were produced. This result was extended to primary and secondary alkylamines, only to find the latter got satisfactory products. It was noticed that the primary alkylamines totally converted to the corresponding imines, which can be reduced by the same reductants.
 | ||
| Scheme 14 Reductive amination of aldehydes and ketones. | ||
The pioneering work for the catalytic reductive alkylation of amines using silanes as reductants was reported by Apodaca and Buchwald. Later in 2005, Yasutaka Ishii and coworkers developed for the first time an iridium complex as a catalyst, which enabled the reductive alkylation of various secondary amines with aldehyde and triethylsilane (Scheme 15).38 In addition to the high selectivity for reduction of imines, it was noteworthy that the reaction underwent smoothly using 1:1:1 molar ratio mixture of amine, aldehyde and Et3SiH. Among the iridium complexes screened, [IrCl(cod)]2 was the most suitable catalyst. Meanwhile, IrCl3 also showed the comparative catalytic activity, which was the synthetic precursor of [IrCl(cod)]2. Similar to Apodaca's research, primary amines were difficult to take place this reaction selectively. PMHS was also examined as the alternative reductant, which gave desired product under the standard conditions. Regarding the mechanism, the authors proposed two plausible pathways, namely the reaction proceeds via the in situ generation of the Ir-dihydride (and/or the Ir-hydride), followed by the reduction of enamine to give the corresponding amine.
 | ||
| Scheme 15 Reductive alkylation of secondary amine. | ||
On-Yi Lee's group also focused on the utilization of ionic hydrogenation in reductive amination of amines, testing other catalysts and organosilanes. In 2008, they reported that with the reaction using InCl3/Et3SiH/MeOH as reagent system, a variety of tertiary amines were constructed in moderate to excellent yields (Scheme 16).39 This nonwater-sensitive reducing reagent system showed remarkable activity for a wide array of substrates, including cyclic, acyclic, aromatic, and aliphatic amines. Functional groups, such as ester, hydroxyl, carboxylic acid, and olefin were well tolerated in the reaction. Besides, the reductive amination of carbonyl compounds resembled a first-order kinetics profile with regard to both InCl3 and Et3SiH. Based on the studies of reaction mechanism employing NMR and ESI-MS, they proposed that the process was initiated by the reaction between InCl3 and MeOH, followed by the transmetalation with Et3SiH to generate [InHCl2(MeOH)x−1], which subsequently transfers the hydride to the iminium ion. With the replacement of MeOH, the desired amine was produced (Fig. 4).
 | ||
| Scheme 16 Reductive amination of carbonyl compounds with secondary amines. | ||
 | ||
| Fig. 4 Proposed reaction mechanism. | ||
Later on in 2009, the same group optimized the previous reagent system by employing Lewis acid as catalyst to expand its applicability in the synthesis of secondary amines (Scheme 17).40 The best results were afforded by use of Fe(II) or Zn(II) complexes as Lewis acids, especially for Zn(ClO4)2·6H2O, which was proven to activate the imine in further research. Control experiments demonstrated that no desired product could be identified when either the Lewis acid or the InCl3 was absent, indicating their necessity in the procedure. The reagent system also exhibited good functional group compatibility and can be applied in the direct reductive amination of various aldehydes and ketones with primary amines without overalkylation products, which didn't go on efficiently to afford the desired amines in previous study. However, aromatic ketones showed unsatisfactory reactivity, proceeding with low conversion.
 | ||
| Scheme 17 Reductive amination of carbonyl compounds with primary amines. | ||
Recently, with the development of ionic hydrogenation, not only the formation of common carbon–nitrogen bond, but also of the N-substituted heterocycles can be accomplished via intramolecular cascade cyclization and ionic hydrogenation as well, where Lewis acid plays key role in both condensation and reduction procedures. The ease of preparation of the nitrogen substituted precursors, the mild reaction conditions, and the efficiency of this reaction that enable cyclization and hydrogenation to be attainable in one step make this protocol an attractive methodology in various kind of heterocycles synthesis.
During studies on the methodology of the construction of heterocycles, Wang and coworkers discovered that Al(OTf)3-catalyzed cascade cyclization and ionic hydrogenation reactions could generate five- and six-membered heterocycles with substitution of various groups in moderate to good yields, including pyrrolidinones, piperidones, and other structure related heterocycles, such as isoindoline, oxazolidinone, and dihydroquinazolinone (Scheme 18).41
 | ||
| Scheme 18 Ionic hydrogenation of nitrogen substituted ketoamides. | ||
Inspired by the previous work on the synthesis of tetralins using TiCl4/Et3SiH as catalyst, they intended to produce pyrrolidinones from N-substituted γ-ketoamides. In the case when 4-oxo-N-phenylpentanamide was employed as substrate with the TiCl4/Et3SiH/CH2Cl2 system, low yield of N-phenyl pyrrolidinone was isolated. To raise the yield of desired product, the reaction conditions were optimized in terms of solvent, Lewis acid and the molar ratio of the reagent system. In their optimization process, the treatment of γ-ketoamide with Al(OTf)3 (0.5 equiv.)/Et3SiH (2 equiv.) in CH3CN and refluxed for 1 h gave access to the anticipated product in highest yield. The reaction not only displayed a broad range of substrate scope, including γ-(δ-)ketoamide, N-aryl amides, carbamates, and open-chain ureas, but also exhibited excellent functional group tolerance, since the electronic effect of the substituents had little influence on the yield of the reaction. Notably, the nitro group substituted ketoamide was compatible with this approach to afford the target product as well, which was considered to be difficult to prepare under conventional conditions. This promising protocol provided a straightforward access to the construction of intramolecular C–N bond, making preparation of structure diverse N-substituted lactams efficiently from a vast array of building blocks under mild reaction conditions.
In order to extend the result to the formation of dihydrobenzohetercycles, which are also important scaffolds due to the high biological activity in natural products and pharmaceutical compounds, the same group went ahead with their research of Al(OTf)3-catalyzed ionic hydrogenation (Scheme 19).42 After optimizing the reaction conditions, 3,4-dihydro-1,4-benzoxazepin-5(2H)-ones and 4,5-dihydro-1,4-benzoxazepin-3(2H)-ones with different substituents were produced smoothly in the utilization of Al(OTf)3/Et3SiH system in good yields. Interestingly, a tricyclic compound and benzodiazepin-5-one were also afforded in moderate yields, which reflected the wide applicability of this approach. Further studies indicated the synthesis of various 3,4-dihydro-1,4-benzoxazines can be achieved successfully by employing N-Boc-2-(2-oxo-2-substituted ethoxy)anilines as the substrates, providing new alternative method in preparing six-membered dihydrobenzoheterocycle. A wide range of functional groups, such as electron-donating groups, electron-withdrawing groups and halides, were well tolerated in all these above reactions. Together with the study in 2013, a facile and versatile Al(OTf)3-mediated cascade cyclization and ionic hydrogenation reaction which allows access to a variety of heterocycles was developed successfully.
 | ||
| Scheme 19 Synthesis of dihydrobenzoheterocycles through ionic hydrogenation. | ||
In the mechanism study, two different reaction pathways are possible: (1) ketone undergoes reduction followed by the amination to form 3,4-dihydro-1,4-benzoxazepin-5(2H)-one, (2) cyclization of ketone occurs prior to the reduction of corresponding imines. Taking 2-(2-hydroxy-2-phenylethoxy)benzamide as the substrate, no target product obtained under standard conditions, while 2-(2-oxo-2-phenylethoxy)benzamide proceeded the reaction availably to yield imine, which was subsequently reduced by Al(OTf)3/Et3SiH to give the desired product. Based on the observations, the authors assumed that the reaction proceeds through a cascade cyclization and reduction pathway in the formation of 1,4-benzoxazepin derivatives (Fig. 5). Simultaneously, similar results were achieved by N-Boc-2-(2-oxo-2-phenylethoxy)aniline.
 | ||
| Fig. 5 Proposed reaction mechanism. | ||
Reduction of CC and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C bond
C bond
It's well acknowledged that varieties of methods have been established for the hydrogenation of unsaturated C–C bonds, where ionic hydrogenation also provides an efficient approach towards the reduction of alkenes and alkynes in mild conditions. Based on the earlier work by Kursanov's group, McKenzie and coworkers reported the stereoselective reduction of indoles substituted with a bulky group at C-3 using the reagent system of Et3SiH–CF3COOH, which gave the cis indolines in moderate yields (Scheme 20).43 Among the reducing agents examined, TES–TFA showed the best selectivity, and was applied in a number of 2-methy1-3-substituted indoles. It is worth noting that 5-fluoro-2-methyl-3-[2-(4-phenylpiperidino)ethyl]-indoline gives access to the corresponding product smoothly as well, which was recovered unchanged from tin and hydrochloric acid. Regarding the stereoselectivity of the TES–TFA reduction, they proposed steric control by the piperazinylethyl side chain probably accounted for the result via a more indoline-like transition state.
 | ||
| Scheme 20 Stereoselective reduction of some indoles. | ||
Later in 2004, to develop a new method for constructing several steroids with unnatural C (20R) configuration, Hazra and coworkers accomplished the highly stereoselective reduction of ketene dithioacetal through ionic hydrogenation, yielding steroidal C (20R) aldehyde in the same reagent system (Scheme 21).44 The author suggested the protonation of ketene dithioacetal occurred first to generate sulfur-stabilised intermediate, followed by the hydrogenation to give the product (Fig. 6). No reduction of the 5,6-double bond took place, proving the chemoselectivity of reduction and indicating the efficiency of ionic hydrogenation in control of stereochemistry.
 | ||
| Scheme 21 Stereoselective reduction of C-20, 22-ketene dithioacetal. | ||
 | ||
| Fig. 6 Proposed reaction mechanism. | ||
In 2003, the utilization of Et3SiH and EtOH in the presence of a catalytic amount of PdCl2 for the reduction of 1-alkenes at room temperature was reported by Mirza-Aghayan and coworkers (Scheme 22).45 With the use of silanes, various long-chain aliphatic olefins were compatible with this reagent system, such as 1-hexadecene and 1-octadecene, giving desired 1-alkanes in high yields. Both of the amount of Et3SiH and reaction time have an influence on the conversion. In addition, the ionic hydrogenation here was kind of exclusive, where no hydrosilylation products were separated.
 | ||
| Scheme 22 Reduction of 1-alkenes to 1-alkanes. | ||
Compared with the advancement of ionic hydrogenation in the reduction of alkenes, the report of such method in the hydrogenation of alkynes was insufficient. Due to the requirement for suitable hydride donors which don't react or react slowly with acid in the alkyne hydrogenation, R. Morris Bullock's group paid attention to the transition metal hydride instead of the Et3SiH. With the reaction using CF3SO3H as the proton donor and Cp(CO)3WH as the hydride donor, several alkynes can be hydrogenated at room temperature in good yields, including phenylacetylene, tert-butylacetylene, bis-(trimethylsilyl)acetylene (Scheme 23).46 The authors proposed that the alkynes were slowly converted to corresponding hydrogenated products via the vinyl triflates intermediates. Notably, the addition of two equivalents of HOTf to the CC bond led to the formation of geminal ditriflate, which was also served as intermediate during the hydrogenation.
 | ||
| Scheme 23 Ionic hydrogenation of alkynes. | ||
Recently in 2013, Katsukiyo Miura and coworkers reported a PtCl2 catalyzed hydrosilylation of terminal alkynes with Et3SiH as the first step of one-pot alkenylation of aldehydes, revealing the significance of ionic hydrogenation in the reduction of CC bond (Scheme 24).47 Oct-1-yne was examined for the methodology, which went through hydrosilylation smoothly at room temperature, giving a mixture of (E)-alkenylsilane and its regioisomer in satisfied yield. Besides, the catalyst system could be applied to other terminal alkynes as well, while a wide range of functional groups were tolerated under the present reaction conditions, such as phenyl, halogen, ester group, etc.
 | ||
| Scheme 24 Hydrosilylation of oct-1-yne. | ||
Reduction of C–X bond
In spite of much progress in the dehalogenation of organic halides, still there are some drawbacks like the toxicity, selectivity and side reaction. More recently, the silicon hydrides has been developed for the removal of halogen substituents, suggesting new approaches in the reduction of C–X bond.In the report of R. Boukherroub's group, the PdCl2-catalyzed reduction of organic halides were carried out in the presence of triethylsilane to afford alkanes in high yields (Scheme 25).48 Various organic halides can proceed ionic hydrogenation availably, and the corresponding products were formed rapidly with high efficiency, while longer reaction time and heating were also indispensable in some cases. A variety of functional groups, such as ester or ether groups, were well tolerated, besides, the substituted patterns had no influence on the outcome of the reaction. Based on the control experiment and GC/MS analysis, an assumed mechanism for the reaction of organic halides through free radicals was anticipated.
 | ||
| Scheme 25 Reduction of organic halides. | ||
In 2004, to overcome the side reaction of NaBH4–InCl3, Akio Baba described an efficient approach for the reduction of alkyl halides at room temperature when Et3SiH–InCl3 was used as the reagent system (Scheme 26).49 The hydrogenation completed in 2 h, producing the satisfied products in moderate to good yields. With the addition of Et3B, the yield of dehalogenation products increased slightly. Nevertheless, as for the less reactive aryl iodide, appropriate InCl3 was also essential in this reaction. Under the conditions employed, the catalytic system was suitable for the radical cyclization of haloalkenes and enynes as well, which succeeded in providing target compounds in moderate yield.
 | ||
| Scheme 26 Reduction of alkyl halides and cyclization of enynes. | ||
An novel and easy practical procedure for the activation and hydrodefluorination (HDF) of C–F bond based on the use of Et3Si[B(C6F5)4] and Et3SiH was developed in 2005 (Scheme 27).50 Varieties of aliphatic halides were compatible for the hydrodefluorination, reflecting the potency of ionic hydrogenation in dehalogenation, such as 1-fluoropentane or even benzotrifluorides. It was noted that the methodology was tolerant of the aryl halide functionality, where the reaction rate was affected by the polarity of solvent, except for the HDF of perfluoroalkanes. For the mechanistic study, the results of GC-MS and 19F NMR suggested that the key to HDF was the abstraction of F− from a C–F bond to generate a carbocation R+, followed by the hydrogenation catalyzed by Et3SiH.
 | ||
| Scheme 27 Hydrodefluorination of aliphatic halides. | ||
Conclusions
Recent advances in ionic hydrogenation of unsaturated organic compounds have been summarized in this review. In addition to the simple reduction of carbonyl group, unsaturated hydrocarbon and imine, reductive amination of carbonyl compounds has also been developed, which provides an attractive alternative to the conventional methods in constructing amines with various functionalities, especially for the development of novel synthesis strategies of diverse heterocycle building blocks via Al(OTf)3-catalyzed cascade cyclization and ionic hydrogenation. Numerous catalytic systems have been developed for the reduction of CO, CN, CC (CC) and C–X bond, such as TFA/Et3SiH, AlCl3/Et3SiH, Bu2SnCl2/PhSiH3 and so on. Therefore, the utilization of hydrogenating pair allows the reactions to proceed more efficiently. Besides, the reaction conditions are generally mild, and a range of functional groups are well tolerated in the process. Although there are much progress in this area, more investigations are still required to extend the substrate scope of ionic hydrogenation, exploring novel and robust catalysts. Furthermore, additional studies are expected to be focused on the development of the asymmetric versions of ionic hydrogenation. In consequence, due to the advantages of ionic hydrogenation, new achievements are expected to appear in the near future.
Acknowledgements
The author gratefully acknowledges the support of the National Natural Science Foundation of China (No. 81473096) and PUMC Youth Fund (No. 3320140011) in producing this review.Notes and references
- A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed.
- C. Hu, D. Creaser, S. Siahrostami, H. Gronbeck, H. Ojagh and M. Skoglundh, Catal. Sci. Technol., 2014, 4, 2427–2444 CAS.
- R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC.
- A. Stolle, T. Gallert, C. Schmoger and B. Ondruschka, RSC Adv., 2013, 3, 2112–2153 RSC.
- L. Hu, L. Lin and S. Liu, Ind. Eng. Chem. Res., 2014, 53, 9969–9978 CrossRef CAS.
- R. Ciriminna, V. Pandarus, F. Béland and M. Pagliaro, Org. Process Res. Dev., 2014, 18, 1110–1115 CrossRef CAS.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- S. Furukawa, Y. Yoshida and T. Komatsu, ACS Catal., 2014, 4, 1441–1450 CrossRef CAS.
- V. Pandarus, G. Gingras, F. Béland, R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2012, 16, 1230–1234 CrossRef CAS.
- E. C. Kleiderer and E. C. Kornfeld, J. Org. Chem., 1948, 13, 455–458 CrossRef CAS.
- H. A. Liebhafsky, J. Am. Chem. Soc., 1937, 59, 452–458 CrossRef CAS.
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef CAS PubMed.
- K. R. Campos, D. Cai, M. Journet, J. J. Kowal, R. D. Larsen and P. J. Reider, J. Org. Chem., 2001, 66, 3634–3635 CrossRef CAS.
- M. Hoogenraad, J. B. van der Linden, A. A. Smith, B. Hughes, A. M. Derrick, L. J. Harris, P. D. Higginson and A. J. Pettman, Org. Process Res. Dev., 2004, 8, 469–476 CrossRef CAS.
- R. A. Grey, G. P. Pez and A. Wallo, J. Am. Chem. Soc., 1981, 103, 7536–7542 CrossRef CAS.
- J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- W. G. Brown, in Organic Reactions, John Wiley & Sons, Inc, 2004, DOI:10.1002/0471264180.or006.10.
- J. V. B. Kanth and M. Periasamy, J. Org. Chem., 1991, 56, 5964–5965 CrossRef CAS.
- J. Halpern, J. F. Harrod and B. R. James, J. Am. Chem. Soc., 1961, 83, 753–754 CrossRef CAS.
- Q. Jiang, Y. Jiang, D. Xiao, P. Cao and X. Zhang, Angew. Chem., Int. Ed., 1998, 37, 1100–1103 CrossRef CAS.
- D. N. Kursanov, Z. N. Parnes and N. M. Loim, Synthesis, 1974, 633–651 CrossRef CAS.
- D. N. Kursanov and N. P. Zinaida, Russ. Chem. Rev., 1969, 38, 812 CrossRef PubMed.
- M. I. Kalinkin, G. D. Kolomnikova, N. P. Zinaida and D. N. Kursanov, Russ. Chem. Rev., 1979, 48, 332 CrossRef PubMed.
- S. Abbina, S. Bian, C. Oian and G. Du, ACS Catal., 2013, 3, 678–684 CrossRef CAS.
- N. Sakai, K. Nagasawa, R. Ikeda, Y. Nakaike and T. Konakahara, Tetrahedron Lett., 2011, 52, 3133–3136 CrossRef CAS PubMed.
- X.-j. Wang, L. Zhang, D. Byrne, L. Nummy, D. Weber, D. Krishnamurthy, N. Yee and C. H. Senanayake, Org. Lett., 2014, 16, 4090–4093 CrossRef CAS PubMed.
- T. Mineno, R. Tsukagoshi, T. Iijima, K. Watanabe, H. Miyashita and H. Yoshimitsu, Tetrahedron Lett., 2014, 55, 3765–3767 CrossRef CAS PubMed.
- N. Sakai, T. Moriya and T. Konakahara, J. Org. Chem., 2007, 72, 5920–5922 CrossRef CAS PubMed.
- N. Sakai, K. Fujii and T. Konakahara, Tetrahedron Lett., 2008, 49, 6873–6875 CrossRef CAS PubMed.
- C. Cheng and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11304–11307 CrossRef CAS PubMed.
- S. Kim, J. Y. Wu, H. Y. Chen and F. DiNinno, Org. Lett., 2003, 5, 685–688 CrossRef CAS PubMed.
- A. J. Cullen and T. Sammakia, Org. Lett., 2004, 6, 3143–3145 CrossRef CAS PubMed.
- G. Li, Q. Xiao, C. Li, X. Wang and D. Yin, Tetrahedron Lett., 2011, 52, 6827–6830 CrossRef CAS PubMed.
- G. Li, L. Li, H. Huang and D. Yin, Org. Biomol. Chem., 2015, 13, 4418–4421 CAS.
- A. C. Fernandes and C. C. Romão, Tetrahedron Lett., 2005, 46, 8881–8883 CrossRef CAS PubMed.
- B. Li, C. B. Bheeter, C. Darcel and P. H. Dixneuf, ACS Catal., 2011, 1, 1221–1224 CrossRef CAS.
- R. Apodaca and W. Xiao, Org. Lett., 2001, 3, 1745–1748 CrossRef CAS PubMed.
- T. Mizuta, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2005, 70, 2195–2199 CrossRef CAS PubMed.
- O.-Y. Lee, K.-L. Law, C.-Y. Ho and D. Yang, J. Org. Chem., 2008, 73, 8829–8837 CrossRef CAS PubMed.
- O.-Y. Lee, K.-L. Law and D. Yang, Org. Lett., 2009, 11, 3302–3305 CrossRef CAS PubMed.
- J. Qi, C. Sun, Y. Tian, X. Wang, G. Li, Q. Xiao and D. Yin, Org. Lett., 2014, 16, 190–192 CrossRef CAS PubMed.
- Y. Tian, X. Wang, Q. Xiao, C. Sun and D. Yin, J. Org. Chem., 2014, 79, 9678–9685 CrossRef CAS PubMed.
- A. E. Lanzilotti, R. Littell, W. J. Fanshawe, T. C. McKenzie and F. M. Lovell, J. Org. Chem., 1979, 44, 4809–4813 CrossRef CAS.
- B. B. Shingate, B. G. Hazra, V. S. Pore, R. G. Gonnade and M. M. Bhadbhade, Chem. Commun., 2004, 2194–2195, 10.1039/b407952c.
- M. Mirza-Aghayan, R. Boukherroub, M. Bolourtchian and M. Hosseini, Tetrahedron Lett., 2003, 44, 4579–4580 CrossRef CAS.
- L. Luan, J.-S. Song and R. M. Bullock, J. Org. Chem., 1995, 60, 7170–7176 CrossRef CAS.
- H. Kinoshita, R. Uemura, D. Fukuda and K. Miura, Org. Lett., 2013, 15, 5538–5541 CrossRef CAS PubMed.
- R. Boukherroub, C. Chatgilialoglu and G. Manuel, Organometallics, 1996, 15, 1508–1510 CrossRef CAS.
- N. Hayashi, I. Shibata and A. Baba, Org. Lett., 2004, 6, 4981–4983 CrossRef CAS PubMed.
- V. J. Scott, R. Çelenligil-Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852–2853 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |