DOI:
10.1039/C5RA15114G
(Paper)
RSC Adv., 2015,
5, 71131-71138
Highly ordered monolithic structures by directional freezing and UV-initiated cryopolymerisation. Evaluation as stationary phases in high performance liquid chromatography
Received
29th July 2015
, Accepted 13th August 2015
First published on 13th August 2015
Abstract
Rigid aligned polymers were prepared by directional freezing and photo-initiated cryopolymerisation. Poly[poly(ethyleneglycol) diacrylate] (polyPEGDA) and poly(butyl methacrylate-co-ethylene glycol dimethacrylate) [poly(BuMA-co-EDMA)] macroporous polymers were prepared by directional freezing of the polymerisation mixture in liquid nitrogen and photo-initiated polymerisation in the frozen state. The polymerisation mixtures consisted of monomer/s (total monomer concentrations > 25 wt%), dioxane as solvent and 2,2-dimethoxy-2-phenylacetophenone as photo-initiator. The porous properties of the macroporous polymers were tuned by modifying the monomer concentration in the polymerisation mixture and the immersion rate. The results obtained suggest that the freezing of the solvent crystals occurs in the direction of the temperature gradient from the surface of the reactor to its centre rather than in the freezing direction. The aligned polymers were prepared in situ within pre-treated cycloolefin copolymer (COC) tubing and the resulting materials were tested as stationary phases for the separation of biomolecules under reversed-phase and hydrophobic interaction chromatography.
Introduction
Directional freezing is a well-known approach for the preparation of both organic and inorganic porous materials having a well ordered porous structure. In this method, a solution of polymer/monomer is frozen in one direction into a cold liquid. During the process, the solvent ice crystals usually grow in the freezing direction and the solidified monomer phase is structured around the aligned solvent crystals which act as template. Since the introduction of the method in 2005,1 two different approaches were developed for the preparation of aligned porous materials. One approach consists of the unidirectional freezing of a solution of pre-formed polymers and the subsequent remotion of the solvent crystals by freeze-drying. Using this method a wide range of aligned polymers were prepared, including hydrophilic and hydrophobic polymers,1–3 sol–gel4 and colloidal solutions.5,6 However, the materials prepared by this approach usually lack good mechanical properties as a result of the lack of covalent bond between the building blocks used (polymer chains, particles, etc.), and this has thus far limited the applications of these materials. In order to overcome these limitations, different research groups have introduced recently a slightly different method in which a solution of monovinyl and/or divinyl monomers is directional frozen, the frozen monomer phase is subsequently polymerised and finally a porous material is obtained by simply removing the solvent by freeze-drying or under vacuum. The polymerisation step can be performed by redox-,7,8 UV-9–11 or gamma-initiated polymerisation.12 The presence of cross-linking points in the polymers prepared by this method greatly improved their mechanical properties and rigidity, which might expand the use of these materials in flow-through applications such as chromatography and solid-phase catalysis. The solvent choice in this method is extremely important. It should have a reasonable high melting point so it remains solid during the polymerisation step. Camphene9 and dioxane9–11,13 with melting points of 50 and 12 °C respectively were used in this synthetic approach. Using this method, Okaji et al.10 prepared honeycomb monolithic polymers using a UV-curable urethane diacrylate monomer (in concentrations between 5 and 10 wt%) and dioxane as solvent. Barrow et al.9 reported the synthesis of methacrylate-based aligned polymers using different monomer concentrations (from around 5 to 20 wt%) and camphene or dioxane as solvents. The prepared polymers showed improved mechanical stability as the monomer concentration increased. The polymer prepared using a monomer concentration of 20 wt% was used as stationary phase for the separation of small molecules in high performance liquid chromatography. The same research group have recently reported the synthesis of temperature and pH-responsive hydrogels using dimethylamino ethyl methacrylate and oligo-ethylene glycol methacrylates as functional monomers.13
For polymer monoliths used as stationary phases in separation science, the majority of polymer-based monoliths explored in chromatography have been based on the same synthetic approach first described by Svec and Fréchet more than 20 years ago.14 This method consists of the preparation of porous monoliths by the free radical polymerisation of a mixture containing a functional monomer and/or cross-linker, radical initiator and a mixture of solvents used as porogens.15 Even though this column technology has recognised advantages over particulate counterparts, it is well established that one of the limiting factors in preparing reproducible polymer monoliths with good chromatographic performance is the degree of bed heterogeneity.16,17 Therefore, alternative polymerisation methods are needed to improve the structural homogeneity of polymer monoliths and allow this column technology to reach the true potential for analytical applications. Directional freezing of a monomer solution followed by cryopolymerisation can potentially overcome these limitations due to the possibility of preparing more homogeneous polymeric scaffolds with controlled porosity.
In this work poly[poly(ethyleneglycol) diacrylate] (polyPEGDA) and poly(butyl methacrylate-co-ethylene glycol dimethacrylate) [poly(BuMA-co-EDMA)] porous monoliths were prepared by directional freezing of the polymerisation mixture and photo-polymerisation using dioxane as solvent. After optimisation of the polymerisation conditions, both polymers were synthesised within modified COC tubing and evaluated as stationary phases for the separation of standard proteins.
Experimental
Materials
Poly(ethyleneglycol) diacrylate Mn ∼ 258 (PEGDA), methyl methacrylate (MMA), butyl methacrylate (BuMA) (99%), ethylene glycol dimethacrylate (EDMA) (98%), ethylene glycol diacrylate (EDA) (90%), glycidyl methacrylate (GMA) (97%), 2-hydroxyethyl methacrylate (HEMA) (97%), 2,2-dimethoxy-2-phenylacetophenone (DMPA) (99%), benzophenone (99%), 2-methyl-4′-(methylthio)-2-morpholinopropiophenone (MTMPP) (98%), phenylbis(2,4,6-trimethylbenzoyl)-phosphine oxide (BAPO) (97%), 4,4′-bis(diethylamino) benzophenone (DEABP) (99%), 1,4 dioxane (99%), basic alumina (Brockman activity I, 60–325 mesh), trifluoroacetic acid (TFA), β-lactoglobulin from bovine milk, ribonuclease A, type I-A, from bovine pancreas, lysozyme from chicken egg white, chymotrypsinogen A, type II, from bovine pancreas and cytochrome C from equine heart were obtained from Sigma-Aldrich (St Louis, MO, USA). PEGDA was purified as described elsewhere.18 All the other monomers were purified by passage through a bed of basic alumina (Brockman activity I, 60–325 mesh) to remove inhibitors. Cycloolefin copolymer (COC) tubing 1 mm i.d. and 2 mm o.d. (Zeonor 1020 R) was supplied by Thermo Scientific (Sunnyvale, CA, USA). Water used in all experiments was purified by a Milli-Q system (Millipore, Bedford, MA, USA).
Instrumentation
An OAI deep UV illumination system (Model LS30/5, San Jose, CA, USA) fitted with a 500 W HgXe-lamp was used for the UV-initiated polymerisation reactions. For calibration, the irradiation power was adjusted to 20.0 mW cm−2, using an OAI model 206 intensity meter with a 260 nm probe head. Mercury intrusion porosimetry studies were performed using a Micromeritics Pore Sizer 9310. The surface morphology of each of the different monoliths prepared in this work was analysed by scanning electron microscopy (SEM) using a FEI Quanta 600 MLA ESEM in the Central Science Laboratory, University of Tasmania. The polymers were sputter-coated with platinum. Chromatographic separations were performed using a Dionex UltiMate™ 3000 HPLC System (Thermo Fisher Scientific, Lane Cove, Australia) equipped with a DGP-3600M gradient pump including a membrane degasser unit, a FLM-3300 column compartment including a capillary flow splitter (split ratio 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) a VWD-3400 UV detector equipped with a 180 nL flow cell and a WPS-3000 autosampler fitted with a 1 μL sample loop. Chromeleon® software (Ver. 6.80) was used for system control and data processing (data collection rate 2.5 Hz).
:1) a VWD-3400 UV detector equipped with a 180 nL flow cell and a WPS-3000 autosampler fitted with a 1 μL sample loop. Chromeleon® software (Ver. 6.80) was used for system control and data processing (data collection rate 2.5 Hz).
Modification of COC tubing
Prior to the preparation of the polymers to be tested as stationary phases, the COC tubing was modified by single step photografting using a modification of the procedure described by Mair et al.19 A 1:1 (wt%) stock solution of MMA and EDA containing benzophenone as photo-initiator (3 wt% respect to monomers) was prepared. This stock solution was deoxygenated with nitrogen for 10 min. The COC tubing were then filled and sealed with specialized fitting. The filled tubing was irradiated under UV light for 12 min. Then, the tubing was rotated 180° and irradiated again for another 12 min. After the photografting reaction, the tubing was washed with methanol and acetone, before drying in a vacuum oven at room temperature overnight.
Preparation of porous monoliths by directional freezing and UV-initiated cryopolymerisation
The monolithic cryopolymers were prepared by free radical polymerisation at −196 °C using a bath of liquid nitrogen and DMPA as photo-initiator (1 wt% respect to monomer). The monomer/s and DMPA were dissolved in 6 g of dioxane. The mixtures used for the polymerisation reactions are presented in Table 1. The mixture was deoxygenated with nitrogen for 10 min. For the polymers to be tested as stationary phases, a 13 cm length COC tubing was filled with the polymerisation mixture and sealed at both ends with appropriate fittings. The filled tubing was attached to a syringe pump (see Fig. 1) and lowered into a bath of liquid nitrogen at different immersion rates (Table 1). After the immersion was completed, the tube with the frozen polymerisation mixture was removed from the cold bath, wiped with a tissue of cold isopropanol (to eliminate cloudiness20) and then irradiated under UV light for 30 min, rotated 180° and irradiated again for another 30 min. In order to avoid melting of the frozen polymerization mixture while it was irradiated, the COC tubing containing the frozen mixture was held over a dewar full of liquid nitrogen (see Fig. 1). After the photo-polymerisation reaction, the tubes with the polymers inside were placed at room temperature to allow melting of the solvent, attached with proper fittings, and flushed with methanol at 120 μL h−1 for 4 h to remove any unreacted components. Bulk polymerisation reactions were carried out in a similar way using NMR tubes as reactor.
Table 1 Polymerisation conditions employed in the preparation of aligned polymer monoliths
| Polymera |
[Monomer] (wt%) |
Immersion rate (cm min−1) |
Median pore diameter (nm) |
Pore volume (mL g−1) |
|
Monomer: cross-linker ratio (wt%) used for poly(BuMA-co-EDMA), poly(GMA-co-EDMA) and poly(HEMA-co-PEGDA) copolymers were 50:50.
|
| Poly(PEGDA)1 |
25 |
|
2321.7 |
1.30 |
| Poly(PEGDA)2 |
25 |
7 |
2641.3 |
1.00 |
| Poly(PEGDA)3 |
30 |
7 |
1381.1 |
0.76 |
| Poly(PEGDA)4 |
35 |
7 |
1067.7 |
0.64 |
| Poly(PEGDA)5 |
25 |
0.7 |
8922.7 |
1.69 |
| Poly(PEGDA)6 |
25 |
0.07 |
22307.3 |
1.96 |
| Poly(BuMA-co-EDMA)1 |
25 |
|
1722.3 |
0.69 |
| Poly(BuMA-co-EDMA)2 |
25 |
7 |
7413.3 |
0.91 |
| Poly(BuMA-co-EDMA)3 |
30 |
7 |
6809.0 |
0.63 |
| Poly(BuMA-co-EDMA)4 |
35 |
7 |
1355.9 |
0.59 |
| Poly(BuMA-co-EDMA)5 |
25 |
0.7 |
13437.7 |
1.92 |
| Poly(BuMA-co-EDMA)6 |
25 |
0.07 |
17838.4 |
1.59 |
| Poly(GMA-co-EDMA)1 |
25 |
7 |
1583.4 |
0.72 |
| Poly(HEMA-co-PEGDA)1 |
25 |
7 |
1769.5 |
1.07 |
 |
| | Fig. 1 Experimental set-up used for the preparation of aligned polymers by directional freezing and UV-initiated cryopolymerisation. | |
Dioxane was removed from these bulk polymers by vacuum drying at 60 °C for 12 h. These materials were later used for mercury intrusion porosimetry analysis.
Chromatographic experiments
Poly(PEGDA) and poly(BuMA-co-EDMA) polymers were used for the separation of standard protein mixtures under hydrophobic interaction chromatography (HIC) and reversed-phase (RP) conditions, respectively. For the separations under RP mode, the eluents used were water containing 0.1% TFA (eluent A) and acetonitrile containing 0.1% TFA (eluent B). For the separations under HIC mode, the mobile phases used were 0.1 M phosphate buffer pH 6.9 containing 3 M ammonium sulphate (eluent C) and 0.1 M phosphate buffer pH 6.9 (eluent D). In all cases the UV absorbance was monitored at 214 nm and each protein concentration was 0.5 mg mL−1 prepared in the starting eluent.
Results and discussion
Preparation of porous monoliths by directional freezing and UV-initiated cryopolymerisation
The commonly found highly ordered structures in aligned macroporous polymers makes of them interesting materials to explore their potential application as stationary phases in liquid chromatography. However, despite considerable improvements in the mechanical properties of these materials achieved by the introduction of the directional freezing and cryopolymerisation approach, the polymer/monomer concentrations commonly used in the polymerisation mixture is relatively low (less than 20 wt%) which tends to form hydrogel-type polymers which can swell or collapse when exposed to different solvents. Porous materials used as stationary phases in HPLC should have good mechanical properties to withstand the high pressures commonly used in the technique and, a rigid porous scaffold to avoid the swelling/collapse of the material when exposed to eluents with different polarity (aqueous buffers and organic solvents). The first focus of this study was to prepare aligned polymer monoliths using higher amounts of monomer (higher than 25 wt%) in the polymerisation mixture. The first attempts consisted on the synthesis of poly(PEGDA) and poly(BuMA-co-EDMA) monoliths from a polymerisation precursor solution containing a total monomer amount of 25 wt% and dioxane as solvent. The polymerisation mixtures were immersed at 7 cm min−1 in the liquid nitrogen bath and the frozen mixture was subsequently irradiated using UV light. SEM images of the polymer prepared by this method are shown in Fig. 2a and b. Aligned monoliths showing a dendritic porous structure were obtained even at monomer concentrations as high as 25 wt%. These porous structures are a replica of the dendritic dioxane crystals formed during the freezing process and similar polymeric scaffolds were previously reported for materials where dioxane was used as solvent.9,10,13
 |
| | Fig. 2 SEM images of poly(PEGDA) (a and c) and poly(BuMA-co-EDMA) (b and d) prepared by directional freezing and frozen photo-polymerisation (a and b) and conventional cryopolymerisation (c and d). | |
In order to compare with the conventional cryopolymerisation technique, the same polymerisation mixture was rapidly immersed into the cold bath and then irradiated under UV light. Poly(PEGDA) (Fig. 2c) and poly(BuMA-co-EDMA) (Fig. 2d) polymers with random porous structure were formed when the polymerisation mixtures were rapidly frozen and then irradiated (conventional cryopolymerisation). These results clearly show the effect of freezing the polymerisation mixture in one direction at a controlled immersion rate.
The use of dioxane as solvent permits the use of a wide range of methacrylate-based monomers bearing different functionality which allows the design of aligned porous polymers with different surface chemistry. As an example, this polymerisation method was used for the preparation of poly(GMA-co-EDMA) polymers (Fig. 3a) bearing reactive epoxide groups which could be further modified by different ring-opening organic chemistry reactions. In the same way, hydrophilic poly(HEMA-co-PEGDA) aligned porous polymers (Fig. 3b) were also prepared. These hydrophilic materials could find applications in processes where biocompatible surfaces are needed (e.g. tissue engineering applications). The polymerisation conditions and porous properties of poly(GMA-co-EDMA) and poly(HEMA-co-PEGDA) monoliths are presented in Table 1. These results demonstrate the versatility of this approach which might be used for the design of ordered porous materials having different chemical functionality (hydrophilic, hydrophobic, reactive).
 |
| | Fig. 3 SEM images of poly(GMA-co-EDMA) (a) and poly(HEMA-co-PEGDA) (b) prepared by directional freezing and frozen photo-polymerisation. | |
All the SEM images shown in this work are of cross-sectional areas of the polymers prepared and therefore, the aligned porous structures obtained are perpendicularly oriented to the freezing direction. This observation is opposite to the commonly reported materials prepared by this method where polymers having aligned pores parallel to the freezing direction were obtained.7,10–12Fig. 4 shows a typical low magnification SEM image of a poly(PEGDA) monolith prepared using a 25 wt% monomer concentration and an immersion rate of 7 cm min−1. Similar low magnification images were observed for all the polymers prepared in this work by directional freezing. These results suggest that the freezing of the dioxane crystals occur in the direction of the temperature gradient (from the surface of the tube to its interior) rather than in the direction in which the tube is immersed within the cold bath. Directional freezing in the direction of the temperature gradient was also reported by Dogu et al.21 for the preparation of organogels with aligned porous structures using cross-linked mixtures of butyl rubber and cyclohexane. The directional freezing in the radial direction of the tubes was explained based in the poor thermal conductivity of the reaction mixture. Even though the prepared materials did not show aligned pores with the freezing direction, the obtained cryopolymers are mechanically stable (due to the high crosslinking density) and showed a considerable more ordered porous structure than previous monolithic cryopolymers prepared in our research group.22,23
 |
| | Fig. 4 SEM image of a cross-sectional area of poly(PEGDA) monolith prepared by directional freezing and UV initiated cryopolymerisation. | |
Effect of monomer concentration
Poly(PEGDA) and poly(BuMA-co-EDMA) porous polymers were prepared from different monomer concentrations including 25, 30 and 35 wt%. FEG-SEM images and pore size distribution curves of the prepared polymers are shown in Fig. 5 and 6. For a comparison, SEM images of the polymers prepared from a 25 wt% monomer concentration are shown in Fig. 2a and b. As the monomer concentration increases more compact structures having smaller median pore sizes were obtained. For the poly(PEGDA) polymers, the median pore size decreased from 2641 nm for poly(PEGDA)2 prepared from a monomer concentration of 25 wt% to 1067 nm for poly(PEGDA)4 prepared from a monomer concentration of 35 wt% (Table 1). A similar trend was observed for poly(BuMA-co-EDMA) porous polymers where the median pore size decreased from 7413 nm for poly(BuMA-co-EDMA)2 prepared from a monomer concentration of 25 wt% to 1355 nm for poly(BuMA-co-EDMA)4 prepared from a monomer concentration of 35 wt%. This observation can be explained by considering the increase in the degree of supercooling of the solvent as the monomer concentration increases.10 An increase in the monomer content in the polymerisation mixture reduces the freezing point of the solvent and consequently the degree of supercooling is higher. This leads to an increase of the instability during the solvent crystal growth and consequently the size of the crystals formed is reduced.
 |
| | Fig. 5 SEM images of poly(PEGDA) (a and b) and poly(BuMA-co-EDMA) (c and d) ordered porous polymers prepared from 30 wt% (a and c) and 35 wt% (b and d) total monomer concentration in the polymerisation mixture. | |
 |
| | Fig. 6 Pore size distribution plots of poly(PEGDA) (upper) and poly(BuMA-co-EDMA) (lower) ordered porous monoliths prepared with different monomer concentrations. | |
Fig. 6 shows bimodal pore size distribution curves for the polymers prepared. These pore size distributions are commonly observed for aligned materials with dendritic pore morphology and it can be explained considering the different processes involved during the solvent crystal formation. During the primary ice crystal formation, the monomer molecules are expelled from the primary solvent crystals and entrapped within channels perpendicularly oriented to the freezing direction.1,24 The pore size distribution curves for the polymers prepared with 35 wt% monomer concentration are broad which might indicate the formation of less regular structures. Similar behaviour was reported for the preparation of poly(hydroxyethyl methacrylate) [poly(HEMA)] hydrogels where the anisotropy of both microstructure and mechanical properties was reduced for hydrogels synthesised with higher monomer concentrations.12
Effect of freezing rate
It is well known that one simple way of modifying the porous structure of aligned polymers is by changing the rate at which the polymerisation mixture is immersed within the cold bath. In this work poly(PEGDA) and poly(BuMA-co-EDMA) monoliths were prepared at 7, 0.7 and 0.07 cm min−1. The monomer concentration of all polymerisation mixtures was fixed at 25 wt%. Fig. 7 and 8 show the SEM images and pore size distribution curves respectively of the resulting polymers. In general, when the freezing rate increases the pore size distribution curves of both poly(PEGDA) and poly(BuMA-co-EDMA) monoliths are shifted towards smaller pore sizes. The median pore size of poly(BuMA-co-EDMA) monoliths was reduced from 17838 for poly(BuMA-co-EDMA)6 to 7413 nm for poly(BuMA-co-EDMA)2, for the cases were the polymerisation mixtures were immersed at 0.07 and 7 cm min−1, respectively (Table 1 and Fig. 8). The same trend was observed for poly(PEGDA) aligned monoliths. This behaviour is related to the fact that increasing the freezing rate the degree of supercooling in the system is higher and consequently the radius of the ice crystal is smaller. For the same reason, it can be explained the broadening of the pore size distribution curves as the immersion rate decreased. These results show that the pore size of these monolithic materials can be controlled by simply adjusting the freezing rate.
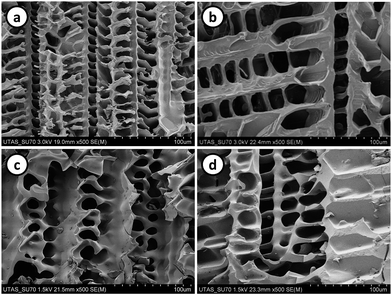 |
| | Fig. 7 SEM images of poly(PEGDA) (a and b) and poly(BuMA-co-EDMA) (c and d) ordered porous polymers prepared at 0.7 (a and c) and 0.07 cm min−1 (b and d). | |
 |
| | Fig. 8 Pore size distribution plots of poly(PEGDA) (upper) and poly(BuMA-co-EDMA) (lower) ordered porous monoliths prepared at different immersion rates. | |
Evaluation of ordered polymer monoliths as stationary phases in HPLC
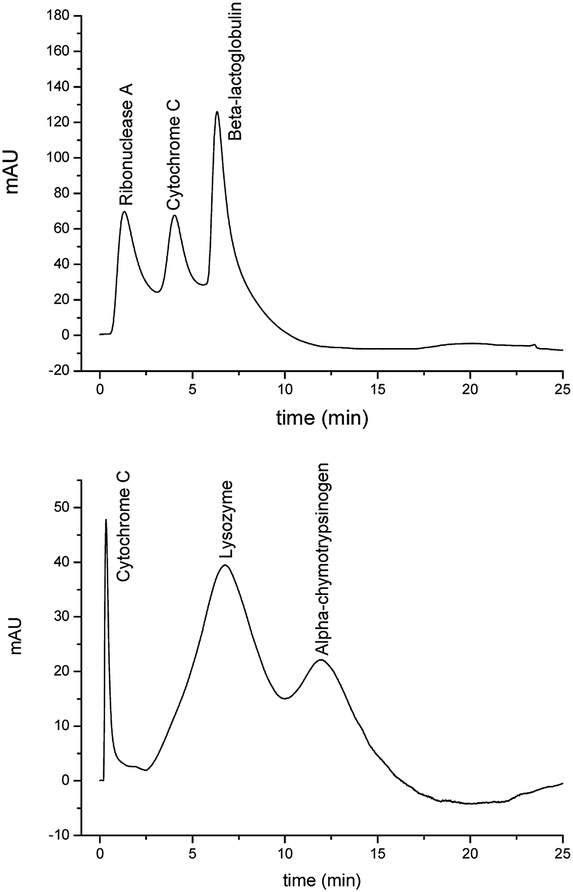
Poly(PEGDA) and poly(BuMA-co-EDMA) monoliths showing a regular porous structure were tested as stationary phases in HPLC. The choice of these two polymers was based in their recognised application as stationary phases in liquid chromatography. The presence of ‘biocompatible’ ethyleneglycol units in the PEGDA structure allowed the use of poly(PEGDA) monoliths as stationary phases under hydrophobic interaction chromatography (HIC).22,23,25 Similarly, the recognised hydrophobicity of the poly(BuMA-co-EDMA) scaffold allowed the application of these materials for reversed phase (RP) separations.26,27 HPLC was chosen as the analytical method for testing these materials because it is a technique where ordered structures should demonstrate a clear advantage and also because it provides an excellent platform to probe the dispersion, diffusion, mass transfer and adsorption properties of these materials. For aligned polymers to be used as stationary phases, the ‘column housing’ chosen as reactor should meet two important requisites. First, it should be ‘transparent’ to UV light in order to ensure a complete photo-initiated polymerisation reaction. Second, its surface should be able to be chemically modified in order to ensure a covalent attachment between the polymer and the tube wall. The last requisite is important to ensure the flow of the mobile phase through the pores of the polymer rather than through the voids between the monolith and the column wall. A material that meets the requisites mentioned above is cycloolefin copolymer (COC) tubing. However, as Flook et al. reported the preparation of polymer monoliths by photo-polymerisation within COC tubing is not a trivial task and the right photo-initiator and wavelength used should be carefully chosen.28 In this study, two different methods were used for the surface modification of COC tubing. The first method consisted in the chemical modification of the COC tubing by hydrogen abstraction using a similar approach to that reported by Flook et al.28 For this, the COC tubing was filled with a 3 wt% solution of MTMPP in t-butanol:water (75:25 v/v) and then irradiated at 260 nm. These modified tubes were then used as ‘moulds’ for the directional freezing and photo-polymerisation reactions. The prepared polymers resulted in poor wall attachment with the polymer coming out of the tubes during the washing steps. BAPO and DEABP were also tested as photo-initiators but these compounds presented limited solubility in the t-butanol:water mixture and the resulting polymers were not attached to the column wall. The second method consisted in the vinylisation of the COC surface by the grafting reaction of a mixture of methyl methacrylate and ethylenediacrylate using benzophenone as initiator.19 The ‘unreacted’ double bounds of the co-polymer layer were later used as anchoring groups of the polymer monolith and the column wall. Although the material was not fully attached to the column wall, the resulting columns withstood the pressures normally used for the polymer washing steps/chromatographic tests without detachment of the polymer from the COC tubing. Based on these results, the latter approach was used for the preparation of poly(PEGDA) and poly(BuMA-co-EDMA) monolithic polymers within COC tubing and subsequently used as stationary phases for the separation of standard proteins. Both polymers were prepared using a 25 wt% monomer concentration and an immersion rate of 7 cm min−1. Fig. 9 shows the separations achieved using poly(BuMA-co-EDMA) and poly(PEGDA) monoliths under reversed phase (RP) and hydrophobic interaction chromatography (HIC) modes, respectively. Separations of the mixtures tested were achieved however with considerable band broadening for all peaks. The separations demonstrated in this case are not as good as those reported by our research group using non-aligned poly(PEGDA) cryopolymers22,23 nor better than that achieved with rigid monoliths prepared by the conventional polymerisation procedures.25,27 Potential reasons for the poorer separations achieved in this work could be due to the fact the polymers are not fully attached to the COC tubing wall as well as the presence of voids observed along the tested columns. Fig. 10 shows optical microscopy images of the polymers within the COC tubes where it is possible to see the presence of empty voids within both columns. The voids in the centre of the column could be due to the formation of solvent crystals of bigger sizes as well as the stress of the material when exposed to drastic temperature changes. These observations might explain the low resolution separations achieved with these monolithic materials. Despite the highly ordered structures observed by SEM, improvements in the attaching procedure between the polymer bed and the column housing are required in order to demonstrate the full potential of these materials as stationary phases for chromatography. Even though the broad peaks observed in Fig. 9 limited the applications of these monoliths as stationary phases in liquid chromatography, similar rigid monoliths having hierarchical porous structure might find applications in other fields such as solid-phase catalysts, drug delivery or as bio-scaffolds for tissue engineering. Moreover, the use of dioxane as solvent of the polymerisation mixture allows the use of high concentrations of metha/acrylate monomers bearing different chemical functionality.
 |
| | Fig. 9 Separations of a protein mixture under RP (upper) and HIC (lower) modes using poly(BuMA-co-EDMA) and poly(PEGDA) polymers, respectively. RP conditions: poly(BuMA-co-EDMA) column length: 11 cm. Flow rate: 65 μL min−1. 1 min isocratic elution with 25% eluent B, followed by a 15 min linear A–B gradient from 25% to 90% B, and then isocratic elution with 90% B for 5 min. HIC conditions: poly(PEGDA) column length: 9.5 cm. Flow rate: 70 μL min−1. 1 min isocratic elution with 100% eluent C, followed by a 15 min linear C–D gradient from 0% to 100% D, and then isocratic elution with 100% D for 5 min. In all cases 1 μL of protein mixture was injected containing 0.5 mg mL−1 each and the UV absorbance was monitored at 214 nm. | |
 |
| | Fig. 10 Optical microscopy images of poly(PEGDA) (left) and poly(BuMA-co-EDMA) (right) polymers within COC tubing. | |
Conclusions
Mechanically stable poly(PEGDA) and poly(BuMA-co-EDMA) monoliths having a regular porous structure were prepared by directional freezing and UV-initiated polymerisation. The use of dioxane as solvent in the polymerisation mixture allowed the synthesis of porous materials with different functionality as well as the use of considerably higher monomer concentrations (up to 35 wt%). The porous properties of the prepared monoliths were tuned by simply modifying the monomer concentration and the immersion rates. The prepared materials were used as stationary phases for the separation of standard proteins under HIC and RP conditions. Even though reasonable good separations were achieved, further work need to be done in order to improve the covalent binding between the monolith and the column wall as well as reduced the formation of voids within the material.
Acknowledgements
This work was supported by the Australian Research Council's Discovery funding scheme (DP130101471). We gratefully acknowledge Dr Karsten Gömann and Dr Sandrin Feig (Central Science Laboratory, University of Tasmania) for assistance with scanning electron microscopy. We also thank Katharina Dihm for assistance with the COC tubing modification.
Notes and references
- H. Zhang, I. Hussain, M. Brust, M. F. Butler, S. P. Rannard and A. I. Cooper, Nat. Mater., 2005, 4, 787–793 CrossRef CAS PubMed.
- H. Zhang and A. I. Cooper, Adv. Mater., 2007, 19, 1529–1533 CrossRef CAS.
- J. W. Kim, K. Taki, S. Nagamine and M. Ohshima, Chem. Eng. Sci., 2008, 63, 3858–3863 CrossRef CAS.
- Y. Minaberry and M. A. Jobbágy, Chem. Mater., 2011, 23, 2327–2332 CrossRef CAS.
- A. Ahmed, R. Clowes, P. Myers and H. Zhang, J. Mater. Chem., 2011, 21, 5753–5763 RSC.
- L. Qian, A. Ahmed, A. Foster, S. P. Rannard, A. I. Cooper and H. F. Zhang, J. Mater. Chem., 2009, 19, 5212–5219 RSC.
- J. Wu, Q. Zhao, J. Sun and Q. Zhou, Soft Matter, 2012, 8, 3620–3626 RSC.
- D. Zhao, J. Zhu, Z. Zhu, G. Song and H. Wang, RSC Adv., 2014, 4, 30308–30314 RSC.
- M. Barrow, A. Eltmimi, A. Ahmed, P. Myers and H. Zhang, J. Mater. Chem., 2012, 22, 11615–11620 RSC.
- R. Okaji, K. Taki, S. Nagamine and M. Ohshima, J. Appl. Polym. Sci., 2012, 125, 2874–2881 CrossRef CAS.
- R. Okaji, K. Taki, S. Nagamine and M. Ohshima, J. Appl. Polym. Sci., 2013, 130, 526–534 CrossRef CAS.
- M. Chen, J. Zhu, G. Qi, C. He and H. Wang, Mater. Lett., 2012, 89, 104–107 CrossRef CAS.
- M. Barrow and H. Zhang, Soft Matter, 2013, 9, 2723–2729 RSC.
- F. Svec and J. M. J. Fréchet, Anal. Chem., 1992, 64, 820–822 CrossRef CAS.
- R. D. Arrua, T. J. Causon and E. F. Hilder, Analyst, 2012, 137, 5179–5189 RSC.
- H. Koku, R. S. Maier, K. J. Czymmek, M. R. Schure and A. M. Lenhoff, J. Chromatogr. A, 2011, 1218, 3466–3475 CrossRef CAS PubMed.
- T. Müllner, A. Zankel, C. Mayrhofer, H. Reingruber, A. Höltzel, Y. Lv, F. Svec and U. Tallarek, Langmuir, 2012, 28, 16733–16737 CrossRef PubMed.
- C. T. Desire, R. D. Arrua, M. Talebi, N. A. Lacher and E. F. Hilder, J. Sep. Sci., 2013, 36, 2782–2792 CrossRef CAS PubMed.
- D. A. Mair, E. Geiger, A. P. Pisano, J. M. J. Fréchet and F. Svec, Lab Chip, 2006, 6, 1346–1354 RSC.
- M. U. Kahveci, Z. Beyazkilic and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4989–4994 CrossRef CAS.
- S. Dogu and O. Okay, Polymer, 2008, 49, 4626–4634 CrossRef CAS.
- R. D. Arrua, P. R. Haddad and E. F. Hilder, J. Chromatogr. A, 2013, 1311, 121–126 CrossRef CAS PubMed.
- R. D. Arrua, A. Nordborg, P. R. Haddad and E. F. Hilder, J. Chromatogr. A, 2013, 1273, 26–33 CrossRef PubMed.
- S. Deville, E. Saiz, R. K. Nalla and A. P. Tomsia, Science, 2006, 311, 515–518 CrossRef CAS PubMed.
- Y. Li, H. Dennis Tolley and M. L. Lee, J. Chromatogr. A, 2010, 1217, 4934–4945 CrossRef CAS PubMed.
- Y. Huo, P. J. Schoenmakers and W. T. Kok, J. Chromatogr. A, 2007, 1175, 81–88 CrossRef CAS PubMed.
- I. Nischang, F. Svec and J. M. J. Fréchet, J. Chromatogr. A, 2009, 1216, 2355–2361 CrossRef CAS PubMed.
- K. Flook, Y. Agroskin and C. Pohl, J. Sep. Sci., 2011, 34, 1–7 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence