
Polyhydroxylated GdDTPA-derivatives as high relaxivity magnetic resonance imaging contrast agents†
Abstract
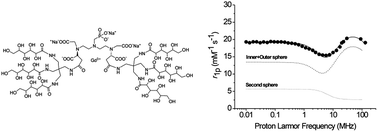
The search for new MRI agents endowed with high relaxivity at the magnetic field strength of clinical scanners (1.5–3 T) is still receiving great attention from researchers involved in the development of new probes. Such Gd(III) complexes should combine a fast inner-sphere water exchange rate (kex) with an enhanced contribution from water molecules in the second and outer coordination spheres. In the present work, dendrimeric-like structures were synthesized by coupling diethylenetriaminepentaacetic acid (DTPA) and its mono-methylphosphonic derivative (P-DTPA) with two differently branched, highly hydrophilic, gluconyl moieties. A 1H and 17O NMR relaxometric study on the corresponding Gd(III) complexes reveals that the Gd-P-DTPA-polyol complex displays very high relaxivities (around 20 mM−1 s−1 at 298 K) over the 0.5–3 T range of field strengths as a result of a fast kex and the presence of a strong second sphere contribution.
- This article is part of the themed collection: RSC Advances Editors' collection: f Block Chemistry
 Please wait while we load your content...
Please wait while we load your content...

