
Electric-field-induced rich magneto-absorption spectra of ABC-stacked trilayer graphene
Yi-Ping Lina,
Chiun-Yan Lina,
Cheng-Pong Changb and
Min-Fa Lin*a
aDepartment of Physics, National Cheng Kung University, 701 Tainan, Taiwan. E-mail: l28981084@mail.ncku.edu.tw; mflin@mail.ncku.edu.tw
bCenter for General Education, Tainan University of Technology, 701 Tainan, Taiwan. E-mail: t00252@mail.tut.edu.tw
First published on 10th September 2015
Abstract
The magneto-optical spectra of ABC-stacked trilayer graphene are enriched by an electric field. A lot of prominent absorption peaks, which arise from the inter-Landau-level transitions, gradually change from twin-peak structures into double-peak ones with increasing electric-field strength. This comes from the destruction in mirror symmetry of the xy-plane and the non-equivalence of two sublattices with identical projections. Specially, a single threshold peak becomes a double-peak structure, owing to the Fermi–Dirac distribution. The absorption frequencies continuously grow or decline except for those caused by the anti-crossings of Landau levels. Also, such anti-crossings can induce extra double-peak structures.
Graphene, with its fascinating hexagonal lattice structure, was first fabricated by mechanical exfoliation in 2004.1 Its unique electronic and optical properties can be easily modulated by changing the layer number,2 the stacking configuration,3 the external magnetic4 and electric fields,5 and the deformed structure.6 These geometric varieties and tuning effects have attracted numerous theoretical and experimental studies especially for the magneto-electronic and -optical properties in few-layer graphene.7–9 The quantized Landau levels (LLs) are strongly dependent on the stacking configuration. N-Layer graphene has N groups of conduction and valence LLs. The three highly symmetric layer configurations, AAA-, ABA-, and ABC-stacked trilayer graphene, exhibit totally different magneto-optical properties. The three categories of excitation channels, belonging to intra-group optical transitions, are revealed in AAA-stacked trilayer graphene. The inter-group LL excitations are absent because of the special relationships among the LL wave functions.10 ABA trilayer graphene is expected to present five categories of absorption peaks since its LLs can be regarded as the superposition of those of the AB-stacked bilayer and monolayer systems. Moreover, ABC-stacked trilayer graphene possesses nine categories of absorption peaks, including all the intra-group and inter-group LL excitations.11 A specific magneto-optical selection rule, Δn = 1 in the variation of quantum number, is identified for the above-mentioned systems. In particular, there are three kinds of the selection rules, Δn = 0, 1, and 2, for sliding graphene with various stacking configurations.12 On the experimental side, only the intra-group excitation channels from the first LL group in AB-stacked few-layer graphenes (N = 1–5) have been verified until now.13 In this work, we focus on how a uniform perpendicular electric field (E = E0ẑ) affects the low-frequency magneto-optical properties of ABC-stacked trilayer graphene, including the structure, intensity and frequency of the absorption peaks.
We have developed a generalized tight-binding model for various external fields,14,15 in which the Hamiltonian matrix is built from the tight-binding basis functions, i.e. the subenvelope functions on the different sublattices. The model can be further utilized to investigate the magneto-optical properties of ABC-stacked trilayer graphene in E. Detailed investigations have been performed on how the inter-LL optical transitions are diversified by the relationship between the interlayer atomic interactions and the electric field. This study shows that the electric field causes the splitting of Landau levels, and hence abundant absorption peaks are induced with E0-dependent peak structure, frequency and intensity. A twin-peak structure or single-peak structure will be replaced by a double-peak structure at a sufficiently large electric-field strength. The extra peaks are generated by the intra-group LL anticrossings during the variation of E0; furthermore, the abnormal E0-dependent absorption frequencies are obtained. The theoretical predictions could be verified by the optical spectroscopy methods.16–19
The magnetic field, electric field and interlayer atomic interactions are simultaneously taken into account in the generalized tight-binding model. The primitive unit cell of ABC trilayer graphene consists of six carbon atoms (A1, B1, A2, B2, A3, B3), where the superscripts 1, 2, and 3 correspond to the first, second, and third layer. The atomic interactions include one intralayer type (β0 = 3.16 eV) and five interlayer types (β0 = 3.16 eV, β1 = 0.36 eV, β2 = −0.01 eV, β3 = 0.32 eV, β4 = 0.03 eV; β5 = 0.013 eV).20 The vector potential caused by a uniform perpendicular magnetic field introduces a periodic phase in the atomic interactions. Thus, the unit cell is enlarged by 2RB times (RB = 79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000/B0), and the Hamiltonian function is a 12RB × 12RB Hermitian matrix. Moreover, a uniform perpendicular electric field leads to a potential energy difference (Vg = E0d; interlayer distance d = 3.35) between two neighboring layers, i.e., Vg changes the site energies of the diagonal Hamiltonian matrix elements. The eigenvalues and eigenfunctions can be solved effectively by a special diagonalization method for a band-like Hamiltonian matrix.
000/B0), and the Hamiltonian function is a 12RB × 12RB Hermitian matrix. Moreover, a uniform perpendicular electric field leads to a potential energy difference (Vg = E0d; interlayer distance d = 3.35) between two neighboring layers, i.e., Vg changes the site energies of the diagonal Hamiltonian matrix elements. The eigenvalues and eigenfunctions can be solved effectively by a special diagonalization method for a band-like Hamiltonian matrix.
The optical absorption function according to the Fermi golden rule at zero temperature is directly obtained through
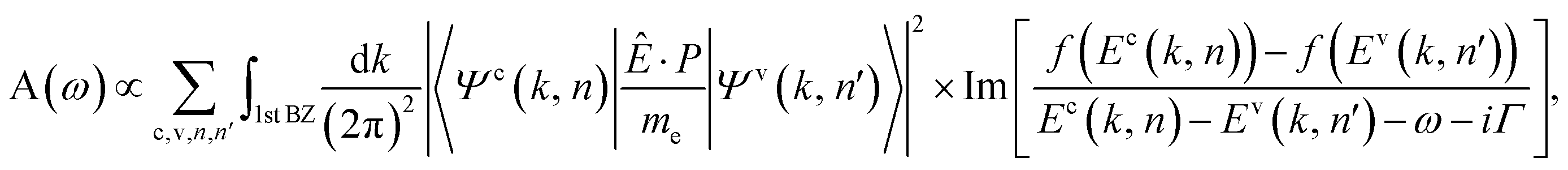
In the presence of an electric field, each LL is twofold degenerate rather than the ordinary fourfold degenerate, mainly owing to the destruction of the xy-plane mirror symmetry. The six subenvelope functions are localized at the 2/6 (5/6; purple) and 4/6 (1/6; blue) positions, as shown in Fig. 1, for B0 = 48 T and Vg = 0.23 eV. The quantum numbers of the first LL group, nc,v1β and nc,v1α, are characterized by the B1j and A3j sublattices, respectively, where the subscripts β and α stand for the localized positions 2/6 and 4/6. Even with the same quantum number, the conduction and the valence LLs have their six subenvelope functions which behave differently. For example, the amplitudes of A1j and A2j (B2j and B3j) in the nv1β = 4 (nc1β = 4) LL are much smaller than those in the nc1β = 4 (nv1β = 4) LL. In general, the subenvelope functions corresponding to the identical planar projections, (A1j ↔ B2j) and (A2j ↔ B3j), are similar in their node numbers and amplitudes. However, the potential difference will destroy the stacking symmetry between these two sublattices, so that the subenvelope functions are no longer equivalent at a sufficiently large Vg. The sharp contrast between the amplitudes of the subenvelope functions is expected to induce dramatic changes in the absorption peaks.
 | ||
| Fig. 1 The first group of Landau levels and their respective six subenvelope wave functions for B0 = 48 T and Vg = 0.23 eV. | ||
The electric field and the atomic interactions lead to a feature-rich energy spectrum, as shown in Fig. 2(a). Some LL energies (Ec,v) monotonously increase or decrease with rising Vg, and the others demonstrate non-monotonic Vg-dependences. In particular, the intra-group LL anticrossings occur frequently during the evolution of the Vg-dependent LL energies (black circles, and green and red rectangles), since some of the LLs possess multi-modes at certain Vg ranges. The amplitudes of the main and side modes in the subenvelope functions are drastically changed.12 For example, the subenvelope functions, (nc1α = 1 & nc1α = 4) and (nv1β = 1 & nv1β = 4), are strongly hybridized around the anticrossing point (Vg ∼ 0.19 eV), the progressions are, respectively, illustrated in Fig. 2(b) and (c) for 0.15 eV ≤ Vg ≤ 0.25 eV. After that, their quantum numbers are exchanged. The modes of the subenvelope wave functions become well-defined again with the further increase of Vg. These variations in the amplitude, waveform, and zero-point number of the subenvelope functions, which are modulated by Vg, directly reflect on the absorption spectra.
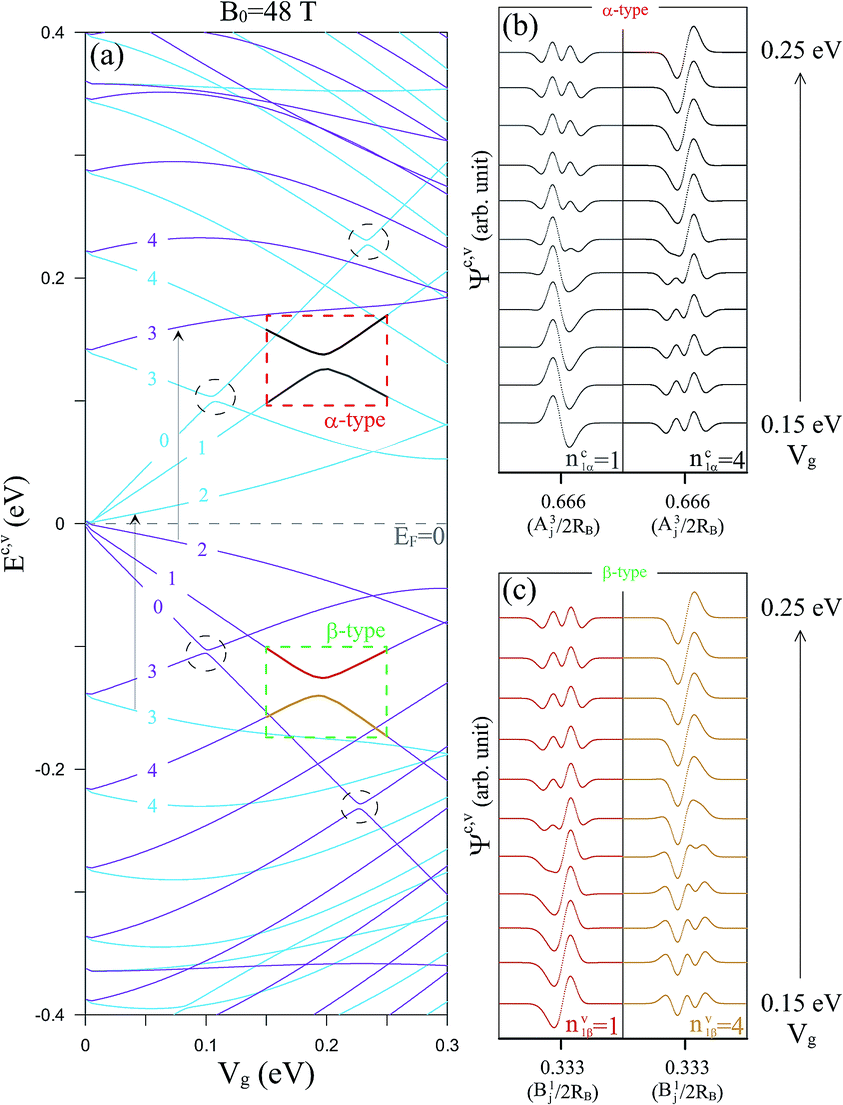 | ||
| Fig. 2 (a) The two Landau-level subgroup spectra at B0 = 48 T as a function of the electric field (Vg) illustrated in two different color codes: purple and blue. The α-type and β-type intra-group Landau-level anticrossings are indicated by red and green rectangles, respectively. (b) The variations of the A3j subenvelope wave functions for ncα = 1 and ncα = 4 with respect to the α-type intra-group Landau-level anticrossing. (c) The variations of the B1j subenvelope wave functions for nvβ = 1 and nvβ = 4 with respect to the β-type intra-group Landau-level anticrossing. | ||
The low-frequency absorption spectra with respect to the first group of LLs exhibit many more special peak structures as a result of being diversified by the electric field. The absorption peaks of the α- and β-type are, respectively, indicated by the blue and purple curves in Fig. 3. The threshold inter-LL excitation, coming from the optical transition: the occupied nv1 = 3 LL to the unoccupied nc1 = 2 LL, exhibits a single-peak structure for Vg = 0, which is attributed to the Fermi–Dirac distribution and the selection rule of Δn = 1.11 The LLs with the different localization positions make the same contribution. The LL degeneracy is destroyed by Vg; therefore, the threshold excitation becomes a double-peak structure. The lower- and higher-frequency comes from nv1β = 2 → nc1β = 3 and nv1α = 3 → nc1α = 2 (arrows in Fig. 2(a)) with their subenvelope functions localized at the 2/6 and 4/6 positions, respectively. The peak structures stays unchanged, and the frequencies increase with the increment of Vg. However, the other absorption peaks for Vg = 0 possess twin-peak structures with a similar absorption intensity,11 which is caused by the asymmetric energy spectrum between the occupied and unoccupied LLs. Such structures also emerge in two different localization positions, 2/6 and 4/6 for a small Vg. As denoted by  and
and  in Fig. 3, where two twin-peak structures or four absorption peaks are displayed at Vg = 0.01 eV. With the further increase of Vg, they are replaced by a double-peak structure (black circle at Vg = 0.11 eV) since a certain peak of the twin-peak structure has almost vanished. In other words, the two peaks in a double-peak structure, respectively, arise from the inter-LL transitions with the distinct localization centers of 2/6 and 4/6. Apparently, the dramatic changes in the peak structure indicate strong competition between the interlayer atomic interactions and the electric field.
in Fig. 3, where two twin-peak structures or four absorption peaks are displayed at Vg = 0.01 eV. With the further increase of Vg, they are replaced by a double-peak structure (black circle at Vg = 0.11 eV) since a certain peak of the twin-peak structure has almost vanished. In other words, the two peaks in a double-peak structure, respectively, arise from the inter-LL transitions with the distinct localization centers of 2/6 and 4/6. Apparently, the dramatic changes in the peak structure indicate strong competition between the interlayer atomic interactions and the electric field.
 | ||
| Fig. 3 The α- and β-type optical spectra in the low-frequency region from Vg = 0.01 to 0.23 eV are shown by the blue and purple colors, respectively. | ||
The Vg-induced rich absorption spectra deserve a closer investigation. The equivalent sublattices, (A1j ↔ B2j) and (A2j ↔ B3j), with identical planar projections, possess similar waveforms and amplitudes for the subenvelope functions.11 The equivalence is destroyed when an electronic static gate voltage induces a potential energy difference on the distinct layers. At a sufficiently large Vg, the amplitudes of A1 and A2 in the valence LLs and those of B2 and B3 in the conduction LLs become very small (red rectangles in Fig. 1). And the spectral intensity of the absorption peak 
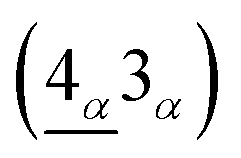 is proportional to the inner product calculated from the Bi sublattices of nvβ = 3 (nvα = 4) and the Ai sublattices of ncβ = 4 (ncα = 3). However, for the absorption peak
is proportional to the inner product calculated from the Bi sublattices of nvβ = 3 (nvα = 4) and the Ai sublattices of ncβ = 4 (ncα = 3). However, for the absorption peak 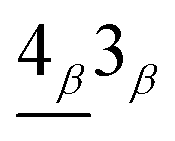
 the intensity is determined by the inner product of the Ai sublattices in nvβ = 4 (nvα = 3) and the Bi sublattices in ncβ = 3 (ncα = 4). As a result, the latter is much weaker than the former.
the intensity is determined by the inner product of the Ai sublattices in nvβ = 4 (nvα = 3) and the Bi sublattices in ncβ = 3 (ncα = 4). As a result, the latter is much weaker than the former.
The electric field induces the intra-group LL anticrossings and thus the extra double-peak structures. The existence of extra absorption peaks is mainly controlled by the weight of the side mode in the progressive LL. For example, the spectral intensities of  and
and  (brown circles in Fig. 3) begin to gradually rise from Vg = 0.15 eV and reach their maximums when Vg gets to 0.19 eV, since the subenvelope functions of nv(c)1β(α) = 1 and nv(c)1β(α) = 4 are strongly hybridized with each other (Fig. 2(b) and (c)). After that, the intensities drop down low under the increasing Vg and disappear when the quantum numbers of the subenvelope functions are restored, becoming well-defined again.
(brown circles in Fig. 3) begin to gradually rise from Vg = 0.15 eV and reach their maximums when Vg gets to 0.19 eV, since the subenvelope functions of nv(c)1β(α) = 1 and nv(c)1β(α) = 4 are strongly hybridized with each other (Fig. 2(b) and (c)). After that, the intensities drop down low under the increasing Vg and disappear when the quantum numbers of the subenvelope functions are restored, becoming well-defined again.
The low-lying inter-LL absorption frequencies (ω![[n with combining low line]](https://www.rsc.org/images/entities/i_char_006e_0332.gif) n) strongly depend on the electric-field strength. The absorption frequencies of the double-peak structures monotonously decrease as Vg increases, while the opposite is true for the threshold double-peak ones, as shown in Fig. 4. Such dependences are revealed to originate from the Vg-dependent LL energies. The perpendicular external electric field induces the opening of the energy gap; therefore, the absolute energies of the six valence and conduction LLs respectively located at the 2/6 and 4/6 positions, which are near the Fermi level and designated to the quantum numbers 0, 1 and 2, will rise with the increasing Vg. However, the behavior of the other LLs at the 2/6 (4/6) position is opposite to these six LLs. As a result, the energy differences of the LLs can be clearly reflected by ωn. Moreover, ωn exhibits some discontinuous Vg-dependent structures at certain electric field strengths (Fig. 4, black circles), mainly owing to the intra-group LL anticrossings. The aforementioned characteristics of the Vg-induced magneto-absorption spectra, like the peak structure, peak intensity, and Vg-dependent absorption frequency, could be verified by optical spectroscopy methods, such as transmission,22,23 reflection24,25 and Raman scattering spectroscopies.16,26,27
n) strongly depend on the electric-field strength. The absorption frequencies of the double-peak structures monotonously decrease as Vg increases, while the opposite is true for the threshold double-peak ones, as shown in Fig. 4. Such dependences are revealed to originate from the Vg-dependent LL energies. The perpendicular external electric field induces the opening of the energy gap; therefore, the absolute energies of the six valence and conduction LLs respectively located at the 2/6 and 4/6 positions, which are near the Fermi level and designated to the quantum numbers 0, 1 and 2, will rise with the increasing Vg. However, the behavior of the other LLs at the 2/6 (4/6) position is opposite to these six LLs. As a result, the energy differences of the LLs can be clearly reflected by ωn. Moreover, ωn exhibits some discontinuous Vg-dependent structures at certain electric field strengths (Fig. 4, black circles), mainly owing to the intra-group LL anticrossings. The aforementioned characteristics of the Vg-induced magneto-absorption spectra, like the peak structure, peak intensity, and Vg-dependent absorption frequency, could be verified by optical spectroscopy methods, such as transmission,22,23 reflection24,25 and Raman scattering spectroscopies.16,26,27
 | ||
| Fig. 4 The Vg-dependent frequencies of the low-lying inter-Landau level absorption peaks. | ||
The generalized tight-binding model was developed to investigate the optical properties of ABC-stacked trilayer graphene in magnetic and electric fields. The prominent LL magneto-absorption peaks are diversified by the electric field. With increased Vg, the twin-peak structures gradually change into the double-peak ones. The former and the latter, which correspond to the inter-LL transitions with the same and different localization centers, are respectively caused by the asymmetric LL energy spectrum and the destruction of both the mirror symmetry and the equivalence of two sublattices. The special peak structures are mainly determined by the competition between the interlayer atomic interactions and the electric-field strength. Especially, a threshold single peak is replaced by a double-peak structure because of the Fermi–Dirac distribution. Moreover, the anti-crossings of the LLs can create extra double-peak structures and thus disrupt the dependence of the peak frequency on Vg. The experimental examinations of the Vg-induced magneto-optical spectra are useful in identifying the stacking configurations among few-layered graphene systems.
References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- J. Nilsson, A. C. Neto, F. Guinea and N. Peres, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 045405 CrossRef.
- M. Koshino, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 125304 CrossRef.
- M. O. Goerbig, Rev. Mod. Phys., 2011, 83, 1193 CrossRef CAS.
- E. V. Castro, K. Novoselov, S. Morozov, N. Peres, J. L. Dos Santos, J. Nilsson, F. Guinea, A. Geim and A. C. Neto, Phys. Rev. Lett., 2007, 99, 216802 CrossRef.
- J. H. Wong, B. R. Wu and M. F. Lin, J. Phys. Chem. C, 2012, 116, 8271 CAS.
- E. McCann and V. I. FalâĂŹko, Phys. Rev. Lett., 2006, 96, 086805 CrossRef.
- T. Morimoto, M. Koshino and H. Aoki, J. Phys.: Conf. Ser., 2013, 012028 CrossRef.
- P. Moon and M. Koshino, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 195458 CrossRef.
- C. W. Chiu, S. C. Chen, Y. C. Huang, F. L. Shyu and M. F. Lin, Appl. Phys. Lett., 2013, 103, 041907 CrossRef PubMed.
- Y. P. Lin, C. Y. Lin, Y. H. Ho, T. N. Do and M. F. Lin, Phys. Chem. Chem. Phys., 2015, 17, 15921 RSC.
- Y. K. Huang, S. C. Chen, Y. H. Ho, C. Y. Lin and M. F. Lin, Sci. Rep., 2014, 4, 7509 CrossRef CAS PubMed.
- S. Berciaud, M. Potemski and C. Faugeras, Nano Lett., 2014, 14, 4548 CrossRef CAS PubMed.
- J. H. Ho, Y. H. Lai, Y. H. Chiu and M. F. Lin, Phys. E, 2008, 40, 1722 CrossRef CAS PubMed.
- S. C. Chen, T. S. Wang, C. H. Lee and M. F. Lin, Phys. Lett. A, 2008, 372, 5999 CrossRef CAS PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235 CrossRef CAS PubMed.
- K. F. Mak, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 104, 176404 CrossRef.
- K. F. Mak, L. Ju, F. Wang and T. F. Heinz, Solid State Commun., 2012, 152, 1341 CrossRef CAS PubMed.
- C. Cong, J. Jung, B. Cao, C. Qiu, X. Shen, A. Ferreira, S. Adam and T. Yu, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 235403 CrossRef.
- J. C. Charlier, J. P. Michenaud and P. h. Lambin, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 4540 CrossRef CAS.
- M. F. Lin and K. W. K. Shung, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17744 CrossRef CAS.
- C. Li, M. T. Cole, W. Lei, K. Qu, K. Ying, Y. Zhang, A. R. Robertson, J. H. Warner, S. Ding and X. Zhang, et al., Adv. Funct. Mater., 2014, 24, 1218–1227 CrossRef CAS PubMed.
- P. Plochocka, C. Faugeras, M. Orlita, M. Sadowski, G. Martinez, M. Potemski, M. Goerbig, J.-N. Fuchs, C. Berger and W. De Heer, Phys. Rev. Lett., 2008, 100, 087401 CrossRef CAS.
- Z. Yan, Y. Liu, L. Ju, Z. Peng, J. Lin, G. Wang, H. Zhou, C. Xiang, E. Samuel and C. Kittrell, et al., Angew. Chem., 2014, 126, 1591 CrossRef PubMed.
- B. Daas, K. Daniels, T. Sudarshan and M. Chandrashekhar, J. Appl. Phys., 2011, 110, 113114 CrossRef PubMed.
- C. Cong, T. Yu, K. Sato, J. Shang, R. Saito, G. F. Dresselhaus and M. S. Dresselhaus, ACS Nano, 2011, 5, 8760–8768 CrossRef CAS PubMed.
- K. Kim, S. Coh, L. Z. Tan, W. Regan, J. M. Yuk, E. Chatterjee, M. Crommie, M. L. Cohen, S. G. Louie and A. Zettl, Phys. Rev. Lett., 2012, 108, 246103 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |