
Controlled aggregation of quantum dot dispersions by added amine bilinkers and effects on hybrid polymer film properties†
Junfeng Yana,
Paul D. McNaughterb,
Ziji Wanga,
Nigel Hodsonc,
Mu Chena,
Zhengxing Cuia,
Paul O'Brienab and
Brian R. Saunders*a
aPolymers, Composites and Carbon Research Group, School of Materials, University of Manchester, Grosvenor Street, Manchester, M9 13PL, UK. E-mail: brian.saunders@manchester.ac.uk
bSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M9 13PL, UK
cBioAFM Facility, University of Manchester, Stopford Building, Oxford Road, Manchester, M13 9PT, UK
First published on 28th October 2015
Abstract
Blends of conjugated polymers with semiconducting nanocrystals (NCs) have attracted much interest as photoactive layers for hybrid polymer solar cells. However, uncontrolled aggregation of the NCs within the hybrid films remains a major processing challenge that adversely affects hybrid film properties and device performance. Here, we deliberately triggered the aggregation of dispersed NCs prior to hybrid polymer film formation and studied aggregation effects on film morphology, thickness and absorbance. The aggregation of parent ZnO and PbS dispersions was triggered by addition of 1,2-ethylenediamine. The latter bilinker is more user-friendly than 1,2-ethanedithiol, which is widely used for NC-based solar cells. The extent of NC aggregation was controlled in this study by the bilinker concentration and mixing time prior to spin coating. The effects of ZnO aggregation on poly(3-hexylthiophene) (P3HT)/ZnO hybrid film morphologies were studied using AFM and SEM. The light absorption and emission properties were probed using UV-visible and photoluminescence spectroscopy. We found that P3HT/ZnO film thickness and absorbance both increased as ZnO aggregation became more pronounced. The results of this study imply that NC aggregation during hybrid polymer solar cell construction may increase light absorption due to increased film thickness. The implications of these effects for hybrid solar cell device optimisation studies are discussed.
Introduction
Semiconducting inorganic nanocrystals (quantum dots) have received considerable attention for use in hybrid polymer solar cells1–8 and quantum dot solar cells.9–12 The former contain nanocrystals (NCs) dispersed within a conjugated polymer matrix and have good potential for future application.1,13 However, there are two key challenges that must be overcome to improve hybrid polymer solar cell power conversion efficiencies.10,14 The first challenge is to bypass residual (insulating) ligands that confer colloidal stability to the NC dispersions used for device fabrication. The second challenge is to overcome uncontrolled NC aggregation.15 By contrast to aqueous nanoparticle dispersions16 colloid stability for non-aqueous dispersions commonly used for NC synthesis (and hybrid solar cell construction) usually relies fully on steric repulsion from ligands. Alivisatos et al. pioneered the replacement of stabilising ligands with small ligands such as pyridine that could be removed using moderate heating.17 However, a consequence of using small ligands is a decrease of colloid stability of the NC dispersions which often results in uncontrolled NC aggregation.1 Whilst ligand exchange using pyridine has been widely used the challenges of forming hybrid polymer films containing well distributed NCs remain.18 Approaches to overcome uncontrolled aggregation include in situ NC growth,19 NC morphology engineering18 and addition of bilinkers after film deposition.14 Bilinkers are ligands that contain two functional groups that can bind to NCs. Despite the importance of aggregation in determining NC nanomorphology within hybrid solar cells there have been very few studies that have examined NC aggregation/morphology relationships. Here, we triggered (and controlled) aggregation of NC dispersions by bilinker addition prior to hybrid film formation. The aims of this study were twofold. Firstly, we aimed to investigate triggered aggregation using a bilinker that was more user-friendly than 1,2-ethanedithiol (EDT). The latter has been used for post-deposition ligand exchange for hybrid polymer/PbS solar cells14 and in NC-based solar cells.11,20 The second aim was to study the effects of NC aggregation on the physical and photoactive properties of hybrid polymer films that have compositions suitable for use in hybrid polymer solar cells.Our previous work established a link between EDT-triggered aggregation of PbS NCs in dispersion and the morphology of spin coated polymer/PbS films.21 However, the hybrid polymer films used for that study contained low volume fractions of NCs and were not capable of being used for hybrid polymer solar cells. In this study we use processing conditions that provide hybrid polymer films with compositions that match those used successfully for hybrid solar cell fabrication elsewhere. It has been shown that post-deposition addition of EDT can improve hybrid solar cell efficiency.14 However, from the application perspective the toxicity, odour and boiling point of a bilinker are important considerations. A bilinker should also have sufficiently low boiling point that it can be removed using moderate heating conditions that do not degrade the conjugated polymer.
In this study we used 1,2-ethylenediamine (EDA) as the main bilinker for triggered (controlled) aggregation prior to film deposition. EDA has a boiling point of 118 °C (cf. 145 °C for EDT) which is close to that of pyridine (115 °C). EDA has a much lower toxicity22 than EDT.23 In addition, EDA does not have the stench hazard that is associated with EDT. EDA is a Lewis base and has several additional advantages compared to EDT when used as a bilinking for NCs. Firstly, amine ligands are known to give weaker trap states than thiols when bound to semiconducting NCs.24 Brewer et al.25 reported that the use of EDA gave much higher charge carrier concentrations and mobilities for deposited Cu2S quantum dot films compared to those prepared using EDT. In addition, amines are less susceptible to the photo-oxidation, which is known to occur for NCs containing thiol-based ligands.26
Here, we studied the controlled aggregation of ZnO dispersions induced by added bilinkers. We also investigated the morphology and optical properties of hybrid polymer films that are candidates for hybrid solar cells. This study focuses on P3HT/ZnO photoactive films (P3HT is poly(3-hexylthiophene)). We selected P3HT because it has been very widely studied in hybrid polymer/NC solar cells1,13,27,28 and is used as a model conjugated polymer. ZnO NCs were primarily used in this study because they do not require long chain stabilising ligands for dispersion stability.29 Furthermore, P3HT/ZnO films are representative candidates for hybrid polymer solar cell construction because P3HT/ZnO solar cells have given efficiencies of 0.7 to 2.0%.13,30
The approach used in this study is shown in Scheme 1A. Concentrated ZnO dispersions were stabilised by added 1-propylamine (1-PA, Scheme 1B) and mixed with P3HT solution containing added bilinker and then spin coated. In this work 1-PA acted as a surfactant and aided ZnO dispersion prior to spin coating. The use of 1-PA to stabilise ZnO dispersions has been well studied in the literature.31–33 By contrast, this study focuses on the effect of added bilinkers on dispersion and hybrid polymer film properties. The bilinkers studied (Scheme 1B) were EDA, EDT and 1,6-hexanediamine (HDA).
 | ||
| Scheme 1 P3HT/ZnO hybrid film preparation method. (A) The ZnO NCs were prepared using 1-PA as surfactant and the bilinker was added to a separate P3HT solution. Subsequently, the two liquids were mixed and after a specific mixing time the dispersion was spin coated to form a film. The composite film contained ZnO NC aggregates that had formed during mixing. (B) Structures of the surfactant and bilinkers used. 1-PA, EDA, EDT and HDA are 1-propylamine, 1,2-ethylenediamine, 1,2-ethanedithiol and 1,6-hexamethylenediamine, respectively. | ||
This study focuses on probing bilinker-structure–morphology relationships for photoactive P3HT/ZnO films. The study does not involve fabrication of solar cells. Here, we first examine the EDA-triggered aggregation of ZnO dispersions and compare the results to those obtained with EDT and HDA to gain insight into the aggregation mechanism. We also demonstrate versatility of EDA as a bilinker using dispersed PbS NCs. Hybrid polymer P3HT/ZnO photoactive layers are then investigated and a morphology phase diagram constructed. The effects of triggered aggregation on the morphology and light absorption behaviours are also studied. Key findings from this study are that aggregation of the NCs increased both the hybrid polymer film thickness and absorbance. Furthermore, these effects were tuneable. It follows that uncontrolled aggregation during hybrid polymer solar cell construction may adversely affect device optimisation.
Experimental
Materials
Zinc acetate dihydrate (98%), EDA (>99.5%), EDT (>98%) and 1,6-hexamethylenediamine (HDA, 98%), hexamethylenediamine (98%) were purchased from Aldrich. 1-Propylamine (1-PA, >99%) was purchased from Alfa-Aesar. Chloroform (99.8%) and methanol (99.8%) were also purchased from Aldrich. The synthesis of the PbS NCs used lead(II) oxide (≥99%, Aldrich), oleic acid (90%, Alfa-Aesar), bis(trimethylsilyl)sulphide (99.9%, Aldrich) and 1-octadecene (90%, Aldrich). P3HT (average molecular weight of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–45000 g mol−1 and 99.995% trace metals basis) was purchased from Aldrich and had a regioregularity >95%. All of the above materials were used as received.
000–45000 g mol−1 and 99.995% trace metals basis) was purchased from Aldrich and had a regioregularity >95%. All of the above materials were used as received.
ZnO NC synthesis
ZnO NCs were synthesised according to the method reported previously.33 The ZnO dispersions were re-dispersed in CHCl3 and 1-PA was gradually added until the 1-PA volume fraction in the co-solvent blend was 13 vol%. The dispersion became transparent after agitation and the ZnO concentration was 18.1 mg mL−1. These ZnO stock dispersions were stored at −18 °C.PbS NC synthesis
PbS NCs were prepared using a modification of the procedure by Hines and Scholes.34 In brief, lead(II) oxide (447.5 mg, 2 mmol) and oleic acid (1.25 mL, 4 mmol) were mixed in 48.5 mL of 1-octadecene under nitrogen. Upon heating to 150 °C for 30 minutes the suspension formed a colourless solution. To this a solution of bis(trimethylsilyl)sulphide (100 μl, 0.5 mmol) in 1-octadecene (15 mL) was rapidly injected. The solution instantaneously turned brown and was heated at 125 °C for 30 s. An excess of acetone was added to the reaction mixture and the NCs were collected by centrifugation at 10000 rpm for 15 minutes. The NCs were re-suspended in 8.0 mL of hexane. Aggregation using acetone, centrifugation and resuspension in hexane was repeated a further two times. The NCs were finally stored in 5.0 mL of hexane under a nitrogen atmosphere.
Preparation of dilute ZnO dispersions
All ZnO dispersion measurements were conducted using CHCl3/1-PA co-solvent blends. Transparent ZnO stock dispersion (1.5 mL) was added to a sample tube and then different volumes of EDA, or EDT (or other ligands as described below), were added and immediately agitated to fully mix the tube contents. The concentrations of bilinker added in this work were described in terms of the mass ratio (MR). The MR value is the ratio of the bilinker mass added to the mass of ZnO present.Hybrid polymer film preparation
The method for depositing P3HT/ZnO films is depicted in Scheme 1A. P3HT (5 mg) was dissolved in CHCl3 (0.5 mL) heated to 60 °C using ultrasonication. Then, the proper volume of bilinker solution was added to give the desired MR. Stock ZnO dispersion (0.5 mL, 18.1 mg mL−1) was then added to the P3HT/EDA solution with rapid manual mixing for specific reaction times (termed “mixing time”) over which bilinker adsorption occurred. Spin coating was performed by adding a portion of the mixed dispersion (0.3 mL) onto pre-cleaned glass substrates (2.5 × 2.5 cm2). The spin coating was conducted using a Laurell, Model WS-650Mz-23NPP spin processor. The latter was programmed to first spin at 300 rpm for 3 s (phase 1) and then at 3000 rpm for another 10 s (phase 2). The bilinkers used appear in Scheme 1B. The deposited P3HT/ZnO films contained a ZnO volume fraction of 28%.Characterisation methods
Dynamic light scattering (DLS) measurements were carried out using a 50 mW He/Ne laser operated at 633 nm with a standard avalanche photodiode and 90° detection optics connected to a Malvern Zetasizer Nano ZS90 autocorrelator. TEM measurements were obtained using a Philips CM20 200 kV instrument (for ZnO) or a Tecnai FEI TF30 (for PbS). Dispersions were prepared and a holey carbon grid used to immerse the solution. SEM was performed using a Zeiss EVO 50 SEM instrument. Energy-dispersive X-ray spectroscopy analysis (EDX) was performed using a Philips XL30 SEM instrument. Optical microscopy was conducted with an Olympus BX41 microscope. Atomic force microscopy (AFM) images were obtained using an Asylum Research MFP-3D operating in AC (“tapping”) mode. Imaging was performed using Olympus high aspect ratio etched silicon probes (OTESPA) with nominal spring constant of 42 N m−1 (Bruker AXS S.A.S, France). Cantilever oscillation frequency varied between 300 and 350 kHz and was determined by the auto-tune facility of the Asylum software, as was the drive amplitude. The set-point was adjusted to just below the point at which tip–sample interaction was lost to minimise sample damage. The thickness of the films was measured using a Dektak 8 (Bruker), Stylus Profilometer. UV-visible spectra were obtained using a Hitachi U-1800 spectrophotometer. The PbS absorption spectrum was measured using a UV-vis-NIR instrument (Perkin-Elmer Lambda 1050) in Wide Beam mode. Photoluminescence spectra were obtained using a double monochromator FLS980 instrument (Edinburgh Instruments, UK) equipped with a xenon lamp and detector (R928P, Hamamatsu). The excitation wavelength was 430 nm.Results and discussion
ZnO nanocrystal and dispersion characterisation
Fig. 1A shows a representative TEM image for the ZnO NCs used in this study. The NCs were well-defined with a narrow size distribution and a number-average diameter from TEM (DTEM) of 4.1 nm (standard deviation of 0.42 nm). The UV-visible absorption spectrum (Fig. 1B) had a wavelength value at half peak height (λ1/2) of 346 nm. From the λ1/2 value and Meulenkamp's equation35 the average size of the ZnO NCs was calculated as 3.5 nm. The latter value is close to the DTEM value.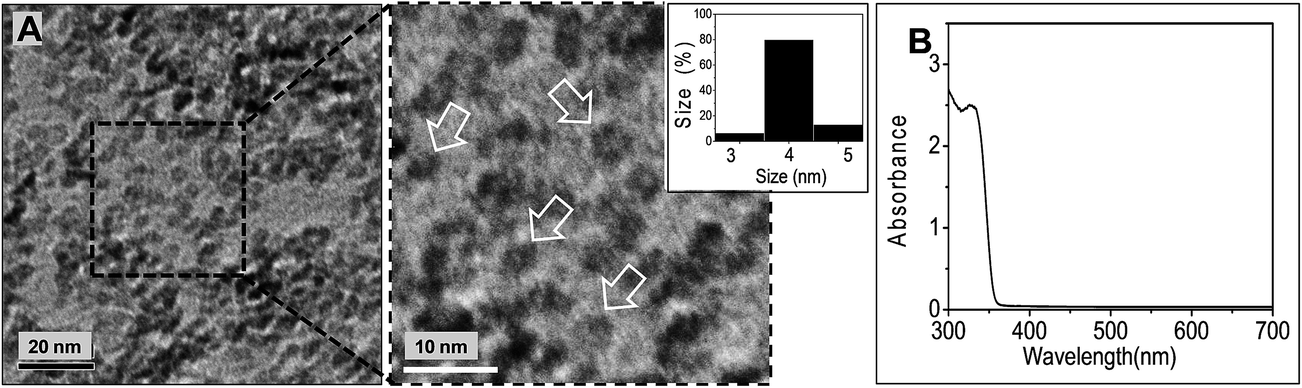 | ||
| Fig. 1 Characterisation of ZnO NCs. (A) TEM image of ZnO NCs deposited from methanol. The inset shows an expanded image and the arrows highlight individual NCs. The size distribution is also shown. (B) UV-visible spectrum for ZnO NCs dispersed in CHCl3/1-PA. | ||
Our aim was to deposit of P3HT/ZnO films using conditions that would translate to hybrid solar cells. Accordingly, it was important to establish conditions where high concentrations of ZnO could be dispersed with minimal aggregation. In a preliminary study we prepared P3HT/ZnO films using ZnO dispersed in CHCl3/methanol blends because this co-solvent blend previously provided good dispersion stability and did not require stabilising ligands.32,33 It is well known that co-solvent blends can improve the performance of polymer-based solar cells.36 Here, the mixed dispersions contained high ZnO and P3HT concentrations in order to deposit photoactive layers with compositions suitable for hybrid solar cells. However, preparation of the concentrated mixed dispersions using CHCl3/methanol blends resulted in large scale ZnO aggregation. To improve dispersion stability prior to P3HT/ZnO film formation we replaced methanol with 1-PA as this stabilising ligand (surfactant) had been used successfully for depositing MDMO-PPV/ZnO hybrid films.32 The use of 1-PA enabled P3HT/ZnO films to be prepared without excessive aggregation (below).
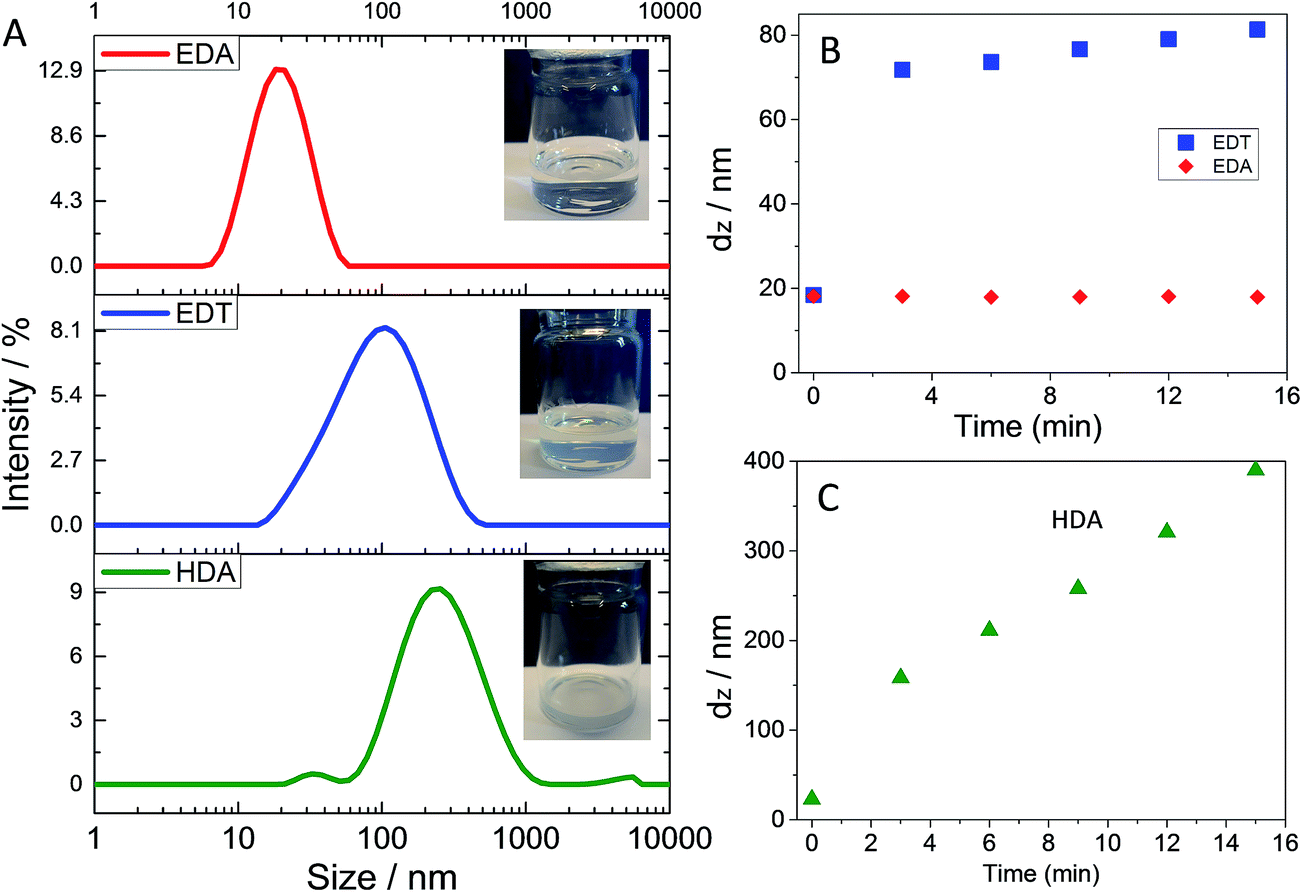
Whilst 1-PA is known to act as a surfactant for ZnO NCs32,33 DLS data have not been reported to support this claim to our knowledge. We therefore studied the effect of 1-PA volume fraction in CHCl3/1-PA solutions on ZnO dispersed particle size using DLS (Fig. 2). With 1-PA volume fractions of 1.5% to 13%, all ZnO solution were transparent. The peak diameter values (Fig. 2A) were in the range 10–20 nm. By contrast the size distribution obtained for ZnO dispersed in pure CHCl3 without added 1-PA (i.e., 0% 1-PA) showed large aggregates. Clearly, 1-PA greatly improved the dispersion of ZnO NCs. Large aggregates were absent for the DLS data when 1-PA volume fractions of 6.5 to 13% were used. The variation of dz with 1-PA volume fraction is shown in Fig. 2B. The lowest dz values (measured for 1-PA volume fractions greater than 6%) were in the range 15–20 nm. The fact that the latter values were greater than the NC sizes measured by TEM (4.1 nm) and UV-visible spectroscopy (3.5 nm) suggests the presence of small aggregates. This limited aggregation is attributed to the short-chain nature of 1-PA. Whilst this ligand was too small to provide complete dispersion of the ZnO NCs, EDA has a low boiling point (48 °C) which should enable its removal from polymer/ZnO films after deposition without excessive heating. It is noted that MDMO-PPV/ZnO hybrid solar cells prepared with added 1-PA had good power conversion efficiencies.32 From Fig. 2A and B the ZnO dispersions were considered to have the best colloidal stability when the EDA volume fraction was in the range of 6.5 to 13 vol%.
 | ||
| Fig. 2 Effect of 1-PA volume fraction in CHCl3/1-PA solutions on dispersed ZnO size. (A) shows the size distributions measured for ZnO dispersions for various 1-PA volume fractions. (B) shows the variation of dz with 1-PA volume fraction. | ||
A strong relationship between ZnO film morphology and ZnO dispersion aggregation was verified by comparing optical and SEM micrographs of spin coating ZnO films from dispersions that contained no added 1-PA or a 1-PA volume fraction of 13%. Images of these dispersions are shown in the insets of Fig. 2B and the spin coated ZnO films are shown in Fig. S1.† Whereas the films spin coated from dispersions that did not contain 1-PA gave poor quality films (Fig. S1A and B†), the film prepared using 1-PA gave a smooth film without micrometre-sized defects (Fig. S1C and D†). The CHCl3/1-PA (13%) was used for preparing ZnO dispersions for the rest of the study.
An important question concerning the ZnO dispersions stabilised by 1-PA is why small aggregates were present. The growth and aging of ZnO dispersions prepared by the sol–gel method have been extensively studied35,37,38 and the particle size increase is generally agreed to occur by an aggregation-based mechanism.37,38 Moreover, the aggregates have been shown to be porous38 and increase in size with increasing ZnO concentration.37 Here, we used high ZnO concentrations (18.1 mg mL−1) to enable P3HT/ZnO films to be spin coated in one step with high volume fractions of ZnO (28 vol%). This approach meant that our films should be capable of being used for hybrid polymer solar cells;13,32 however, it also led to limited aggregation. To test the effect of stabilising ligand structure 1-hexanethiol (1-HT) was added to a ZnO dispersion stabilised by 1-PA. This dispersion had been stored at room temperature to increase aggregate size. The addition of 1-HT rapidly decreased the dz value from 27 to 15 nm (see Fig. S2†). This result suggests that thiol ligands bind more strongly to ZnO than primary amines and that the aggregates were partially reversible. The 1-HT ligand has a much higher boiling point (152 °C) than 1-PA (48 °C) and films containing 1-HT would require higher temperature for removal with a greater chance of polymer degradation. Consequently, we used 1-PA as the stabiliser in this study. The concentrated ZnO dispersions studied here contained small aggregates which we term primary aggregates and they were partially reversible based on the results from 1-HT study (Fig. S2†) described above.
Controlled-aggregation of NC dispersions by addition of bilinkers
Having established conditions where 1-PA improved ZnO dispersion stability we next explored the ability of added EDA to trigger aggregation. DLS data are shown in Fig. 3A as a function of MR. Aggregation of the dispersions was demonstrated for EDA when the MR reached 0.1 because the distribution moved to larger sizes and larger aggregates were detected. We term these aggregates, secondary aggregates. This term is taken to apply to aggregated clusters of primary aggregates. Visually, the transparent dispersions became turbid at MR = 0.1 and strongly turbid when MR = 0.2 (see Fig. 3B). Fig. 3C shows that as MR increased, the dz value strongly increased once MR reached 0.10. Accordingly, the critical MR value for secondary aggregate formation of ZnO triggered by added EDA was identified as 0.10. This is the first report of triggered aggregation of ZnO dispersions by EDA to our knowledge and shows that triggered aggregation is not limited to EDT.29 | ||
| Fig. 3 Triggered aggregation of ZnO dispersions by added EDA. (A) shows DLS data obtained at different MR values. The data were obtained 6 min after addition of EDA. (B) shows images of the tubes for different MR values. (C) shows the variation of dz (from the data shown in (A)) with MR. (D) Proposed mechanism for bridging of two ZnO surfaces by EDA after partial ligand exchange with 1-PA (see text). | ||
What is the mechanism for EDA-triggered aggregation? Because ZnO NCs dispersed in non-polar organic solvents have very low effective Hamaker constants15,29 the two-particle attractive van der Waals interaction is weak. Adsorbed short-chain ligands (e.g., 1-PA) provide short-range (steric) repulsive interactions. By contrast, EDA bilinkers that bridge neighbouring ZnO surfaces provide attractive interactions (via amine-to-Zn2+ coordinate bonds). Because primary amines do not bind strongly to ZnO we assume that ligand exchange of bound 1-PA with other ligands (e.g., EDA) is rapid. When two nearby ZnO surfaces containing EDA adsorbed in monodentate manner collide, ligand exchange with 1-PA could occur which would then enable bridging of the surfaces by EDA. The total interaction from van der Waals attraction and bridging EDA bilinkers and can be thought of as an inter-NC bond if attraction is stronger than repulsion. Accordingly, total interaction should change from repulsive to attractive with increasing number of bridging EDA bilinkers and MR (see Fig. 3D). In this way large secondary aggregates could build up and their size should continually increase as a function of time (below). If the above mechanism is correct then aggregation should become more pronounced if either the strength of the binding or the bilinker length is increased. To test these ideas we investigated ZnO dispersion aggregation using EDT or HDA as bilinkers because they were expected to bind more strongly to ZnO or were longer than EDA (Scheme 1B).
The abilities of EDA, HDA and EDT to trigger aggregation of the ZnO dispersions were compared using a MR of 0.02 and a mixing time of 6 min (see Fig. 4). This MR value was selected because it was less than the critical value of 0.10 for EDA-triggered aggregation (above). By contrast to EDA, addition of EDT caused pronounced aggregation as evidenced by the large peak at ∼100 nm (Fig. 4A). Fig. 4B shows that for EDT addition the dz value increased rapidly in the first 2 min and then linearly with time. These data confirm the trend noted above using 1-HT that thiol groups have stronger binding ability than amino groups for ZnO. This conclusion is congruent with the work of Chen et al.39 where it was shown that the binding of octadecanethiol was stronger than that of dodecylamine for ZnO. Primary amines and thiols are hard and soft bases, respectively, in terms of the Hard Soft Acid Base theory.40,41 However, Zn2+ is a borderline acid.40,41 We propose from the results presented here that ZnO prefers to bind to soft bases.
 | ||
| Fig. 4 Comparison of various bilinker ligands for triggered ZnO aggregation. (A) shows DLS size distributions obtained for the dispersions at 6 min after addition of blinker ligand using MR = 0.02. The insets show images of the tubes. (B) and (C) show dz measured at various times after addition of the bilinkers. | ||
An interesting finding from the data shown in Fig. 4 is that HDA caused strong aggregation (Fig. 4A and C) and this occurred for an MR value where EDA did not cause aggregation. It follows that the longer HDA chain was more effective at either displacing 1-PA ligands or directly bridging neighbouring NCs under the conditions employed. It is not clear how HDA would increase 1-PA exchange. Consequently, a mechanism for triggered aggregation of ZnO involving direct bridging of ZnO surfaces by bilinkers which span both surfaces with monodentate binding to each is favoured (see Fig. 3D).
To test the generality of EDA as a bilinker the ability of EDA to trigger aggregation of PbS NCs was investigated. PbS NCs are also promising candidates for hybrid solar cells.14,42 The dispersed PbS NCs were stabilised by oleic acid and had a size of about 4 nm as judged by TEM (Fig. S3A†). The PbS NCs absorbed light strongly and had an absorption maximum at 1170 nm (Fig. S3B†). Fig. 5A and B shows the effect of added EDA and EDT, respectively, on the size distributions for PbS. In both cases aggregates became evident with increasing mixing time. The data imply that EDA exchanged with some of the oleic acid ligands which triggered aggregation. However, this effect was relatively slow for EDA (Fig. 5A) compared to EDT (Fig. 5B). The latter was more effective removing oleic acid as judged by the shift of the whole peak to large sizes. This difference indicates that PbS also has a greater affinity for thiol groups than amine groups. For both ZnO and PbS, EDA is able to trigger aggregation; albeit, at a slower rate than EDT. Because EDA and EDT are much smaller than oleic acid these results imply that the mechanism for PbS aggregation involved ligand exchange followed by aggregation. It is proposed that the bilinkers removed oleic acid (and decreased steric repulsion) by ligand exchange and this process enabled the bilinkers to bridge neighbouring NCs once the NCs were able to move sufficiently close for bridging to occur.
 | ||
| Fig. 5 Triggered aggregation of PbS dispersions byadded bilinkers. DLS data are shown for added (A) EDA or (B) EDT. The MR value used for these measurements was 0.1 and the mixing times are shown. | ||
We can now summarise our proposed mechanism for EDA-triggered NC aggregation. There appears to be a competition between binding sites on the NCs for ligand binding groups and this is governed by the binding affinities for the surface. Unidentate amine (for ZnO/1-PA) or carboxylate (for PbS/oleic acid) bound stabilising ligands can be displaced by thiol or amine bilinkers and this process leads to aggregation. EDA binding was weaker for both ZnO and PbS compared to EDT because of weaker primary amine affinity for those surfaces. Aggregation was enhanced by bilinkers that are long compared to the stabilising ligands present.
Effects of aggregation on P3HT/ZnO film properties
There are three photoactive layer properties that strongly affect hybrid polymer solar cell power conversion efficiency15 that were investigated in this study; morphology at the nanometre scale, film thickness and light absorption. We first investigated the effect of triggered aggregation on the morphology of spin coated P3HT/ZnO films. These films are representative photoactive layers for hybrid solar cells30,43–46 and contained a ZnO volume fraction of 28%. To begin the film study optical micrographs were used to construct a phase diagram for the films (Fig. 6). The film prepared using ZnO stabilised by 1-PA (without added bilinker) can be seen from the MR = 0 image after 5 min mixing time in Fig. 6 (top left hand image). There was no evidence of micrometre-scale aggregation. The effect of added EDA on the morphology of P3HT/ZnO films was investigated (Scheme 1A) and Fig. 6 shows optical microscopy images of the films obtained using various MR values and mixing times. Films prepared using low MR values and mixing times had a smooth appearance with few, if any, micrometre-sized features evident. However, films prepared using high MR values and longer mixing times exhibited many micrometre-sized aggregates. We refer to films without and with micrometre-scale aggregates as 1-phase and 2-phase films, respectively. The MR value for the appearance of micrometre-sized aggregates was ca. 0.15 for mixing times of 3 and 5 min. For a mixing time of 1 min the MR value for the films to show micrometre-sized aggregates was 0.30. | ||
| Fig. 6 Morphology phase diagram for P3HT/ZnO films. The MR and mixing times for EDA-triggered aggregation are shown. The closed and open circles represent 2-phase and 1-phase structures on the micrometre-scale. Scale bars = 10 μm. | ||
In order to probe the effect of added EDA at the nanometre-scale morphology the films were studied using AFM and SEM. The effect of MR at a constant mixing time (1.0 min) was first investigated (Fig. 7) (larger area AFM images from the same samples with line profiles are shown in Fig. S4†). The P3HT/ZnO film prepared in the absence of added bilinker exhibited undulations (Fig. 7A) with an RMS surface roughness of 5.6 nm and a maximum height difference of 20 nm. These values show that our preparation conditions (for MR = 0) gave films that were smoother than comparable films reported in the literature.13,27 Furthermore, the morphology for our MR = 0 film and its roughness are similar to those reported by Beek et al.32 for their MDMO-PPV/ZnO films which contained 26 vol% ZnO. By contrast P3HT/ZnO films prepared in the absence of 1-PA exhibited pronounced phase separation with many micrometre-scale aggregates present (see Fig. S5†) and were not suitable for further study.
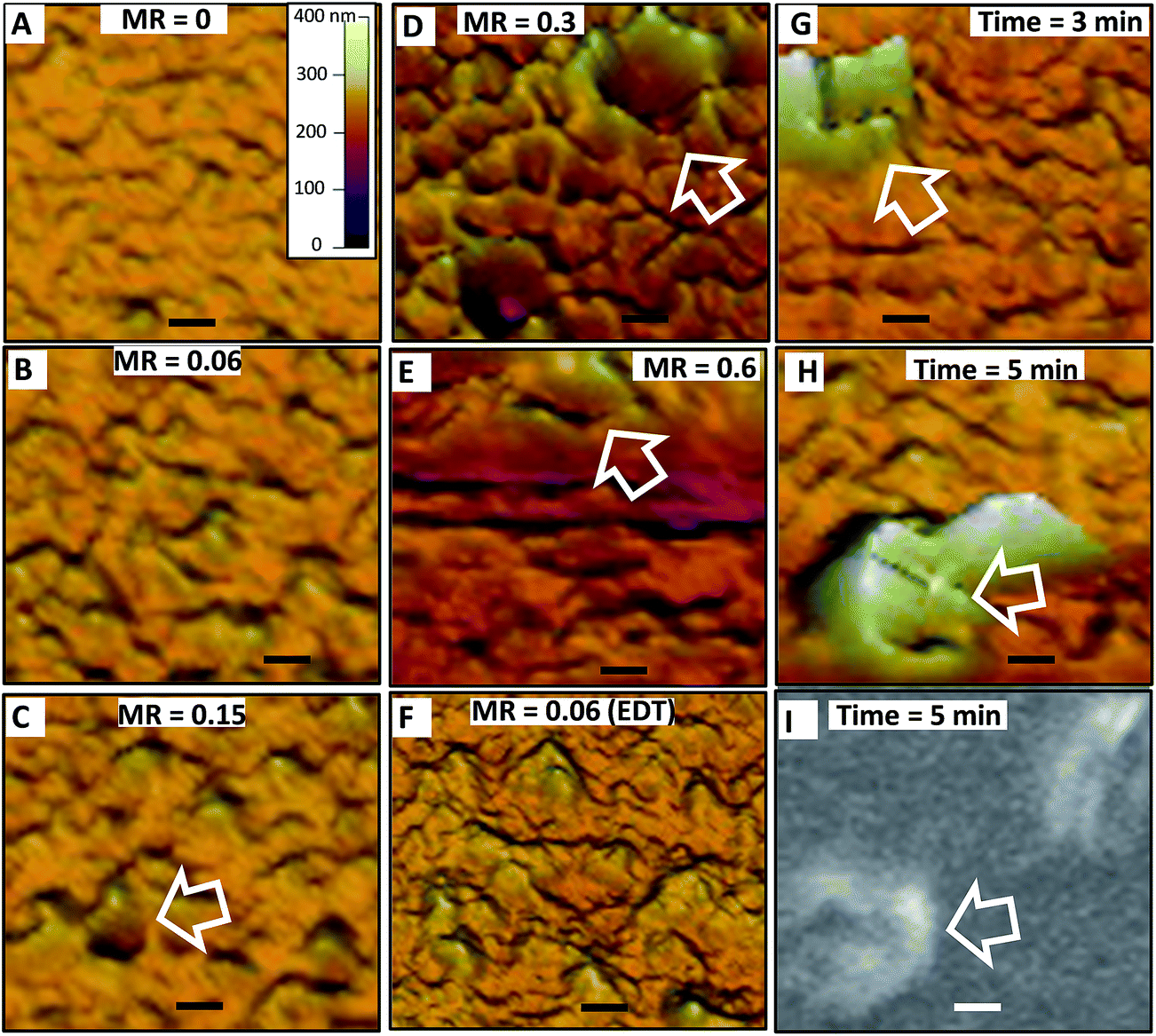 | ||
| Fig. 7 Effect of mass ratio and mixing time on the morphology of P3HT/ZnO films. AFM images as a function of MR (A–F) for a 1 min mixing time. (G) to (I) show AFM and SEM images at different mixing times (MR = 0.15). The scale in (A) applies to all AFM images. The scale bars are 500 nm. The arrows identify crater-like aggregates (see text). | ||
Having established conditions that provided smooth films we then investigated the effect of added bilinker on morphology. The AFM images shown in Fig. 7B–F (and line profiles in Fig. S4F†) show the effects of MR on morphology for a 1 min mixing time. The smooth undulations present at low MR values (Fig. 7B) became increasingly pronounced as MR was increased to 0.15 and occasional crater-like features became evident (Fig. 7C–E). The undulations and crater-like features increased in size as MR increased from 0.3 to 0.6 (Fig. 7D and E). These features are be due to micrometre-scale phase separation and are attributed EDA triggered-aggregation of ZnO that occurred in dispersion prior to film formation. Fig. 7F shows an AFM image for a film when EDT had been added (MR = 0.06). The undulations present for that system are comparable to those evident for the equivalent film prepared using EDA (Fig. 7B). The line profiles obtained from larger area measurements for the P3HT/ZnO films (Fig. S4A–E†) and RMS roughness as a function of MR are shown in Fig. S4F and G.† These data confirm the trends noted from the surface morphologies above. The film prepared using MR = 0.6 film was not investigated using AFM because of excessive phase separation present (see Fig. 6); however, crater-like features were present as judged by SEM data (Fig. S6,† inset).
The effect of mixing time at constant MR (of 0.15) on the surface morphology was also investigated (Fig. 7G and H) (note that Fig. 7C corresponds to 1 min mixing time and should also be considered for the present discussion). As the mixing time increased the undulations and crater-like features increased. To confirm that the later features were not an artefact of AFM, an SEM images were obtained (Fig. 7I and S6†). The differences in contrast from the SEM images suggest that the walls of these crater-like features were rich in ZnO. This suggestion was supported by selected area EDX measurements (Fig. S6†) which showed Zn/S elemental ratios of 2.7 and 1.8, respectively, for a crater-like aggregate and a crater-like aggregate-free film section. Hence, P3HT/ZnO film morphology was strongly controlled by ZnO aggregation in the dispersed phase prior to film deposition and this became increasingly pronounced with increasing MR and mixing time. When MR values greater than 0.1 were used micrometre-sized aggregates became evident from the DLS data for the parent dispersions (see Fig. 3A). Consequently, it is likely that micrometer-sized aggregates formed in dispersion were then deposited, and coated to some extent by P3HT, to give the morphologies apparent in Fig. 7G to I.
The thickness of the P3HT/ZnO films increased both with increasing MR (see Fig. 8A) or mixing time (Fig. 8B). These effects, which resulted from ZnO, were unexpected since the most of the film (72 vol%) was P3HT. We propose that the film thickness increase is due to triggered aggregation of the ZnO dispersion prior to spin casting for the following reasons. Firstly, our previous work29 established that EDT-triggered aggregation of concentrated ZnO dispersions by EDT formed macroscopic physical gels. In that work triggered aggregation increased dispersion viscosity to the point where the fluid no longer flowed under its own weight. Furthermore, high resolution SEM and TEM images showed that space-filling networks of ZnO NCs were present.29 Because concentrated ZnO dispersions were used in the present study to prepare the hybrid films it follows that the viscosity of the mixed dispersions increased as triggered aggregation became more pronounced (as MR or mixing time increased). It is also well known that the thickness of spin coated films increases with increasing viscosity47,48 because of increasing resistance to radial flow. Consequently, the behaviours evident in Fig. 8 are attributed to viscosity increases caused by triggered aggregation of the ZnO component. Thus, NC aggregation increased the thickness of hybrid polymer photoactive layers. This result has important implications for hybrid polymer photoactive layers where uncontrolled aggregation is often problematic.15 It follows from our results that uncontrolled aggregation prior to film deposition will lead to an increase in film thickness. Moreover, differences in local aggregation may also result in inhomogeneous thicknesses for the film.
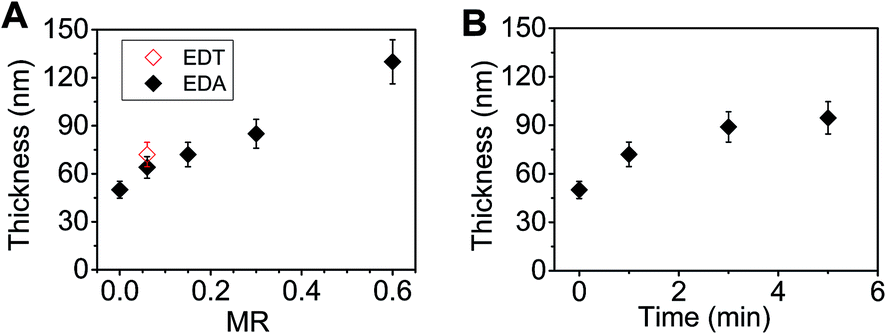 | ||
| Fig. 8 Effect of preparation conditions P3HT/ZnO film thickness. The effects of (A) MR (for a mixing time of 1 min) and (B) mixing time (for a MR value of 0.15) on film thickness are shown. | ||
Effects of triggered aggregation on P3HT/ZnO film light absorption
UV-visible spectra were measured for the films to probe the effects of triggered aggregation on the absorbance properties. Both the effect of MR (Fig. 9A and B) and mixing time (Fig. 9C and D) were investigated. The spectra (Fig. 9A and C) show strong absorption from P3HT vibronic bands in the 500 to 600 nm region.49 The maximum P3HT absorbance increased with increasing MR value (Fig. 9B). A data point for a P3HT/ZnO film prepared using added EDT (MR = 0.06) is also shown. The latter film gave a slightly increased P3HT absorption compared to the film prepared using EDA. The effect of mixing time prior to film deposition was also investigated using MR = 0.15 (see Fig. 9C and D). It can be seen from Fig. 9D that increased mixing time also increased the absorption from P3HT, which is due to more extensive aggregation. The results shown in Fig. 9 demonstrate clearly that the absorption of P3HT is strongly affected by aggregation of dispersed ZnO prior to film formation. As aggregation became more pronounced, so did the extent of light absorption. These results are new for hybrid polymer films and imply that aggregation of the NCs during film deposition will increase the absorption from the polymer phase. This could be disadvantageous if precise control of absorption was required, which is often the case for device optimisation studies and of course scale up. | ||
| Fig. 9 Effect of mass ratio and mixing time on the light absorption of P3HT/ZnO films. (A) UV-visible spectra for various MR values (mixing time 1 min). (B) Variation of absorbance at 550 nm with MR. A data point obtained using added EDT is also shown. (C) UV-visible spectra for different mixing times (MR = 0.15). (D) Variation of absorbance with mixing time. | ||
Why did the absorption of P3HT increase as the aggregation of dispersed ZnO became more pronounced? To examine the causes of the latter effect the variation of absorbance was plotted as a function of thickness for the films (see Fig. 10) (only data obtained using MR less than 0.6 were included because those data did not show evidence of excessive light scattering). The data show linear behaviour. We estimated the absorption coefficient of P3HT (αP3HT) starting from the simple Beer–Lambert law,50
| I(x) = I(0)e−αx | (1) |
| I(t) = I(0)10−A | (2) |
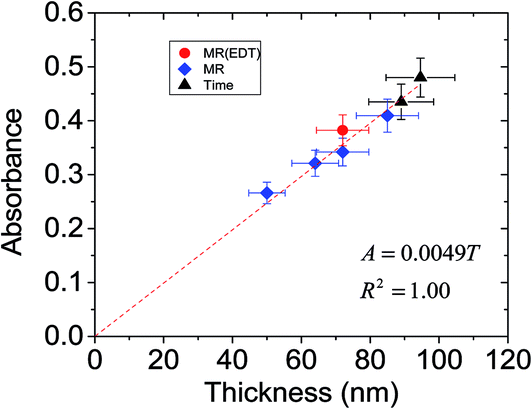 | ||
| Fig. 10 Relationship between absorbance and P3HT/ZnO film thickness. These data are from Fig. 8 and 9. | ||
For a film thickness, x = T, eqn (1) and (2) can be equated. Furthermore, in the wavelength range of 500 to 600 nm light absorption resulted from P3HT, which had a volume fraction of ϕP3HT. Accordingly, to a first approximation the value for the P3HT absorption coefficient (αP3HT) can be calculated from eqn (3).
 | (3) |
It follows from eqn (3) that the absorbance is proportional to thickness and the gradient from a plot of absorbance vs. thickness will be equal to (αP3HTϕP3HT)/2.303. Fig. 10 shows that good linearity was obtained between absorbance and thickness. Using a ϕP3HT value of 0.72 and the gradient from Fig. 10 we estimate αP3HT = 1.6 × 105 cm−1. This value is reasonably close to the αP3HT value of ∼2.5 × 105 cm−1 reported by Kim et al. for their P3HT films.51 Moreover, the data clearly show that it is the increase of film thickness associated with increased ZnO aggregation that is responsible for the absorbance increase with MR and mixing time evident in Fig. 9.
To probe the effects of aggregation on charge transfer across the P3HT/ZnO interface we measured the photoluminescence (PL) spectra for a P3HT/ZnO film deposited using MR = 0.15 as well as spectra for control P3HT and P3HT/ZnO film (Fig. S7†). The latter film was prepared in the absence of added EDA (MR = 0). The spectra were corrected for the optical density of the films. The PL spectra, which were dominated by the vibronic transitions of P3HT,52 showed increased quenching in the order P3HT/ZnO (MR = 0) > P3HT/ZnO (MR = 0.15) > P3HT. It follows from these data and the morphology discussion above that triggered aggregation due to EDA (for the film prepared using MR = 0.15) decreased the ZnO/P3HT interfacial area and, hence, decreased charge transfer due to increased exciton recombination. Hybrid polymer solar cells with photoactive layers affected by excessive exciton recombination are likely to have decreased current density and power conversion efficiencies.15
Conclusions
In this study we have examined the effects of controlled (triggered) aggregation of ZnO dispersions and its effects on the morphology, thickness and light absorption of photoactive P3HT/ZnO films. Spin coating conditions were employed that gave films with a NC volume fraction suitable for use in hybrid polymer solar cells. We introduced a new bilinker EDA that is less toxic and does not have the pungent odour of EDT. Our results show that EDA can be used to trigger aggregation of ZnO dispersions stabilised by 1-PA. EDA could also trigger the aggregation of oleic acid stabilised PbS NCs, albeit at a slower rate, showing the effect is general. However, the aggregation rate is system dependent. Furthermore, we have shown that aggregation of ZnO dispersions caused an increase in P3HT/ZnO film thickness. Moreover, the increase in thickness also increased the light absorption of the films due to P3HT. These results have general importance for construction of hybrid polymer solar cells because the mixed dispersions used often have marginal or insufficient colloid stability as a consequence of compromises being made between ligand length and insulating effect of ligands in the final film. Our results suggest that uncontrolled aggregation will lead to the inability to precisely control film thickness and absorption, which would adversely affect device optimisation studies for hybrid polymer solar cells especially when formulation composition is varied. For P3HT/ZnO films, aggregation may also lead to decreased charge transfer across the P3HT/ZnO interface.Acknowledgements
BRS and POB would like to thank the EPSRC for funding this work (K010298/1). We also thank Edinburgh Instruments (UK) for their help with obtaining the PL data.References
- J. Yan and B. R. Saunders, RSC Adv., 2014, 4, 43286–43314 RSC.
- F. Alam, N. Kumar and V. Dutta, Org. Electron., 2015, 22, 44–50 CrossRef CAS.
- S. Yao, Z. Chen, F. Li, B. Xu, J. Song, L. Yan, G. Jin, S. Wen, C. Wang, B. Yang and W. Tian, ACS Appl. Mater. Interfaces, 2015, 7, 7146–7152 CAS.
- C. Leow, T. Harada, T. Ohnishi and M. Matsumura, RSC Adv., 2015, 5, 22647–22653 RSC.
- C. Giansante, R. Mastria, G. Lerario, L. Moretti, I. Kriegel, F. Scotognella, G. Lanzani, S. Carallo, M. Esposito, M. Biasiucci, A. Rizzo and G. Gigli, Adv. Funct. Mater., 2015, 25, 111–119 CrossRef CAS.
- Z. Li, W. Wang, N. C. Greenham and C. R. McNeill, Phys. Chem. Chem. Phys., 2014, 16, 25684–25693 RSC.
- A. J. MacLachlan, T. Rath, U. B. Cappel, S. A. Dowland, H. Amenitsch, A. C. Knall, C. Buchmaier, G. Trimmel, J. Nelson and S. A. Haque, Adv. Funct. Mater., 2015, 25, 409–420 CrossRef CAS PubMed.
- W. Fu, L. Wang, Y. Zhang, R. Ma, L. Zuo, J. Mai, T. K. Lau, S. Du, X. Lu, M. Shi, H. Li and H. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19154–19160 CAS.
- K. S. Jeong, J. Tang, H. Liu, J. Kim, A. W. Schaefer, K. Kemp, L. Levina, X. Wang, S. Hoogland, R. Debnath, L. Brzozowski, E. H. Sargent and J. B. Asbury, ACS Nano, 2012, 6, 89–99 CrossRef CAS PubMed.
- L.-Y. Chang, R. R. Lunt, P. R. Brown, V. Bulović and M. G. Bawendi, Nano Lett., 2013, 13, 994–999 CrossRef CAS PubMed.
- E. J. D. Klem, D. D. MacNeil, L. Levina and E. H. Sargent, Adv. Mater., 2008, 20, 3433–3439 CrossRef CAS.
- G. H. Carey, A. L. Abdelhady, Z. Ning, S. M. Thon, O. M. Bakr and E. H. Sargent, Chem. Rev., 2015 DOI:10.1021/acs.chemrev.5b00063.
- F. Li, Y. Du and Y. Chen, Thin Solid Films, 2012, 526, 120–126 CrossRef CAS.
- J. Seo, M. J. Cho, D. Lee, A. N. Cartwright and P. N. Prasad, Adv. Mater., 2011, 23, 3984–3988 CrossRef CAS PubMed.
- B. R. Saunders and M. L. Turner, Adv. Colloid Interface Sci., 2008, 138, 1–23 CrossRef CAS PubMed.
- S. Chen, D. A. MacLaren, R. T. Baker, J. N. Chapman, S. Lee, D. J. Cole-Hamilton and P. Andre, J. Mater. Chem., 2011, 21, 3646–3654 RSC.
- W. U. Huynh, J. J. Dittmer, W. C. Libby, G. L. Whiting and A. P. Alivisatos, Adv. Funct. Mater., 2003, 13, 73–79 CrossRef CAS.
- I. Gur, N. A. Fromer and A. P. Alivisatos, J. Phys. Chem. B, 2006, 110, 25543–25546 CrossRef CAS PubMed.
- S. Dowland, T. Lutz, A. Ward, S. P. King, A. Sudlow, M. S. Hill, K. C. Molloy and S. A. Haque, Adv. Mater., 2011, 23, 2739–2744 CrossRef CAS PubMed.
- J. M. Luther, M. Law, Q. Song, C. L. Perkins, M. C. Beard and A. J. Nozik, ACS Nano, 2008, 2, 271–280 CrossRef CAS PubMed.
- R. Rhodes, P. O'Brien and B. R. Saunders, J. Colloid Interface Sci., 2011, 358, 151–159 CrossRef CAS PubMed.
- Sigma-Aldrich Safety Data Sheet, Ethylenediamine, Product Number 03550, http://www.sigmaaldrich.com, accessed April 2015.
- Sigma-Aldrich Safety Data Sheet, 1,2-Ethanedithiol, Product Number 02390, http://www.sigmaaldrich.com, accessed April 2015.
- D. A. Hines and P. V. Kamat, J. Phys. Chem. C, 2013, 117, 14418–14426 CAS.
- A. S. Brewer and M. S. Arnold, Thin Solid Films, 2014, 567, 91–95 CrossRef CAS.
- W. Wang, A. Kapur, X. Ji, M. Safi, G. Palui, V. Palomo, P. E. Dawson and H. Mattoussi, J. Am. Chem. Soc., 2015, 137, 5438–5451 CrossRef CAS PubMed.
- Y. Shi, F. Li and Y. Chen, New J. Chem., 2013, 37, 236–244 RSC.
- S. D. Oosterhout, M. M. Wienk, M. Al-Hashimi, M. Heeney and R. A. J. Janssen, J. Phys. Chem. C, 2011, 115, 18901–18908 CAS.
- A. Mispelon, J. Yan, A. H. Milani, M. Chen, W. Wang, P. O'Brien and B. R. Saunders, RSC Adv., 2015, 5, 18565–18577 RSC.
- S. D. Oosterhout, M. M. Wienk, S. S. van Bavel, R. Thiedmann, L. J. Koster, J. Gilot, J. Loos, V. Schmidt and R. A. Janssen, Nat. Mater., 2009, 8, 818–824 CrossRef CAS PubMed.
- S. Alem, J. Lu, R. Movileanu, T. Kololuoma, A. Dadvand and Y. Tao, Org. Electron., 2014, 15, 1035–1042 CrossRef CAS.
- W. J. E. Beek, M. M. Wienk, M. Kemerink, X. Yang and R. A. J. Janssen, J. Phys. Chem. B, 2005, 2005, 9505–9516 CrossRef PubMed.
- R. Rhodes, M. Horie, H. Chen, Z. Wang, M. L. Turner and B. R. Saunders, J. Colloid Interface Sci., 2010, 344, 261–271 CrossRef CAS PubMed.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
- E. A. Meulenkamp, J. Phys. Chem. B, 1998, 102, 5566–5572 CrossRef CAS.
- J. Griffin, A. J. Pearson, N. W. Scarratt, T. Wang, A. D. F. Dunbar, H. Yi, A. Iraqi, A. R. Buckley and D. G. Lidzey, Org. Electron., 2015, 21, 216–222 CrossRef CAS.
- L. Spanhel and M. A. Anderson, J. Am. Chem. Soc., 1991, 113, 2826–2833 CrossRef CAS.
- M. S. Tokumoto, S. H. Pulcinelli, C. V. Santilli and A. F. Craievich, J. Non-Cryst. Solids, 1999, 247, 176–182 CrossRef CAS.
- J. Chen, R. E. Ruther, Y. Tan, L. M. Bishop and R. J. Hamers, Langmuir, 2012, 28, 10437–10445 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- T.-L. Ho, Chem. Rev., 1975, 75, 1–20 CrossRef CAS.
- A. Watt, T. Eichmann, H. Rubinsztein-Dunlop and P. Meredith, Appl. Phys. Lett., 2005, 87, 253109 CrossRef.
- N. C. Das and P. E. Sokol, Renewable Energy, 2010, 35, 2683–2688 CrossRef CAS.
- S. R. Ferreira, R. J. Davis, Y.-J. Lee, P. Lu and J. W. P. Hsu, Org. Electron., 2011, 12, 1258–1263 CrossRef CAS.
- F. Li, W. Chen, K. Yuan and Y. Chen, Org. Electron., 2012, 13, 2757–2762 CrossRef CAS.
- T. C. Monson, M. T. Lloyd, D. C. Olson, Y.-J. Lee and J. W. P. Hsu, Adv. Mater., 2008, 20, 4755–4759 CrossRef CAS.
- P. Jiang and M. J. McFarland, J. Am. Chem. Soc., 2004, 126, 13778–13786 CrossRef CAS PubMed.
- S. Middleman, J. Appl. Phys., 1987, 62, 2530–2532 CrossRef.
- C. Hellmann, N. D. Treat, A. D. Scaccabarozzi, J. Razzell Hollis, F. D. Fleischli, J. H. Bannock, J. de Mello, J. J. Michels, J.-S. Kim and N. Stingelin, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 304–310 CrossRef CAS.
- J. N. Nelson, The physics of solar cells, Imperial College Press, London, 2003 Search PubMed.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch, C.-S. Ha and M. Ree, Nat. Mater., 2006, 5, 197–203 CrossRef CAS.
- M. Baghgar and M. D. Barnes, ACS Nano, 2015, 9, 7105–7112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra15009d |
| This journal is © The Royal Society of Chemistry 2015 |