
Influence of carbon nanomaterial defects on the formation of protein corona†
 a
a
Abstract
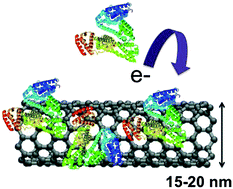
In any physiological media, carbon nanomaterials (CNM) strongly interact with biomolecules leading to the formation of biocorona, which subsequently dictate the physiological response and the fate of CNMs. Defects in CNMs play an important role not only in material properties but also in the determination of how materials interact at the nano-bio interface. In this article, we probed the influence of defect-induced hydrophilicity on the biocorona formation using micro-Raman, photoluminescence, infrared spectroscopy, electrochemistry, and molecular dynamics simulations. Our results show that the interaction of proteins (albumin and fibrinogen) with CNMs is strongly influenced by charge-transfer between them, inducing protein unfolding which enhances conformational entropy and higher protein adsorption.
 Please wait while we load your content...
Please wait while we load your content...

