
CFD simulations of catalytic hydrodeoxygenation of bio-oil using Pt/Al2O3 in a fixed bed reactor
Abstract
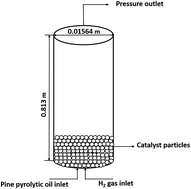
The upgradation of pyrolysis bio-oil by a hydrodeoxygenation (HDO) process using a Pt/Al2O3 catalyst is numerically investigated using a computational fluid dynamics (CFD) based commercial solver, ANSYS Fluent 14.5. In the simulations, a fixed bed reactor with concurrent upflow of unprocessed bio-oil and H2 gas is introduced. The dimensions of the reactor and the thermo-physical properties of all three phases are adopted from experimental data available in the literature. The size of the catalyst particles is 10 μm and a loading of 60 g is used in the present simulations. The main aim of this work is to numerically investigate the effects of weight hourly space velocity (WHSV), temperature (T) and pressure (P) on the upgradation of bio-oil using catalytic hydrodeoxygenation and to delineate the effects of pertinent parameters on the distribution of the various products of the upgraded biofuel. For this purpose, the following ranges of parameters are considered in this work: weight hourly space velocity, WHSV = 2–4 h−1, temperature, T = 623–673 K, and pressure, P = 6996–10 443 kPa. Briefly, the results indicate that a lower WHSV and higher pressure are favourable for the yield of aromatics and the conversion of high non-volatile components, however the effect of temperature is negligible. Finally, a comparison is made between the conversion of high non-volatiles and phenols and the yield of alkanes and aromatics obtained in fixed bed and fluidized reactors for identical values of WHSV, temperature and pressure. It is found that the performance of the HDO of bio-oil using a Pt/Al2O3 catalyst in a fixed bed reactor is superior in all aspects in comparison to fluid bed reactors in the present range of pertinent parameters.
 Please wait while we load your content...
Please wait while we load your content...

