
Optimizing Wnt-3a and R-spondin1 concentrations for stem cell renewal and differentiation in intestinal organoids using a gradient-forming microdevice†
Asad A. Ahmada,
Yuli Wangb,
Christopher E. Simsb,
Scott T. Magnessc and
Nancy L. Allbritton*ab
aDepartment of Biomedical Engineering, University of North Carolina, North Carolina State University, Chapel Hill, NC 27599, Raleigh, NC 27695. E-mail: nlallbri@unc.edu; Fax: +1 919 962 2388; Tel: +1 919 966 2291
bDepartment of Chemistry, University of North Carolina, Chapel Hill, NC 27599, USA
cDepartment of Medicine, Division of Gastroenterology and Hepatology, University of North Carolina, Chapel Hill, NC 27599, USA
First published on 27th August 2015
Abstract
Epithelial renewal in the colonic crypt is a tightly controlled process and essential for normal digestive function. Crypts contain both proliferative and differentiated cells with the balance orchestrated by the interplay of proliferation and differentiation signals. While critically important to homeostatic renewal, the threshold concentrations of factors such as Wnt-3a and R-spondin1 that promote stem cell renewal are unknown. A simple, linear gradient-generating device was used to screen a wide range of Wnt-3a and R-spondin1 concentrations for their impact on a large number of colon organoids (colonoids) to achieve statistically significant results while minimizing the amounts of costly reagents and primary tissue samples. Matrigel-embedded colonoids were tracked for the presence of stem/transit-amplifying cells, differentiated cells and overall colonoid size in response to varying concentrations of Wnt-3a and R-spondin1, presented singly and in combination. A Wnt-3a concentration of 60 ng mL−1 and R-spondin1 concentration of 88 ng mL−1 were identified as the critical concentrations required for stem-cell renewal and colonoid expansion. At these concentrations, the colonoids possessed similar viability, size and passage efficiency compared to colonoids cultured under standard growth concentrations. The gradient culture system enabled efficient determination of the minimal growth factor concentrations needed to produce a physiologically-relevant colonoid phenotype. Relative to traditional culture conditions, lower factor concentrations yielded the added benefit of a more morphologically appropriate colonoid possessing columnar cells surrounding a central lumen with active crypt-like bud formation.
Introduction
The health of the colon is partly dependent on the chemical milieu surrounding the colonic stem-cell niche. The epithelial monolayer lining the colon is supplied by constantly renewing cells migrating from the stem-cell niche. This niche resides at the base of colonic crypts, local invaginations that harbor the intestinal stem cells, their immediate progeny and supporting cells.1 The stem cells give rise to transit-amplifying cells that proliferate and differentiate into absorptive colonocytes, mucus-producing goblet cells, and hormone-producing enteroendocrine cells. These non-dividing cells migrate to the luminal surface where they undergo apoptosis and exfoliate. This process drives complete replacement of the colonic epithelium every 4–7 days making this tissue the most actively self-renewing tissue in the body.2 Stem-cell self-renewal and differentiation are known to be modulated by the interplay of intrinsic gradients of mitogens, morphogens, and differentiation factors.3 Much remains to be understood as to how these various chemical factors control the process of epithelial homeostasis in health and disease including their threshold concentrations for activity. To better study these processes, in vitro model systems that enable precise control of the stem-cell environment are needed.In vitro models of cell proliferation and differentiation in the colon have been hampered by the inability to recapitulate the key features of normal intestinal epithelial tissue. Most studies have been restricted to in vivo inspection, histological assessment, or cancer cell lines that are incapable of normal differentiation.4,5 Recent advances in organotypic culture techniques now enable the culture of primary human and mouse stem cells derived from colonic crypt tissue.1,6–8 Under specific culture conditions, both single intestinal stem cells and isolated crypts grow into self-organizing, functional 3D epithelial organoids or “mini-guts” containing stem cells and the full repertoire of differentiated colonic epithelial cell types.2,9–11 When these in vitro cultured mini-guts are derived from colon tissue they are referred to as colonoids. Colonoid culture requires explicit growth conditions in which the cells are suspended in Matrigel, a 3D laminin and collagen-rich matrix similar to the basal lamina propria, which is further supplemented with a mixture of growth factors including Wnt-3a, R-spondin1, epidermal growth factor (EGF), noggin, and jagged. These conditions maintain stem-cell multipotency and enable culture of the colonoids for greater than 1 year while maintaining a normal karyotype.1,12 Colonoids generated either from isolated crypts or individual stem cells grow into cystic structures with multiple crypt-like buds projecting outward from a central lumen.13 Remarkably, cell-renewal kinetics, differentiation and crypt patterning characteristics recapitulate those seen in vivo. Stem cells in the colonoids give rise to transit-amplifying cells that proliferate, differentiate, undergo apoptosis and are shed into the central lumen 3–5 days later.12,14 This 3D culture system has enabled a rapidly growing number of studies elucidating molecular mechanisms involved in stem-cell renewal and differentiation, membrane transport, intestinal regeneration, and carcinogenesis.10,15,16
In vitro cell-based screens using these types of primary organotypic tissue mimics are poised to greatly improve our understanding of the biological effects of intrinsic and extrinsic factors on cell renewal and physiology.14,16 The combination of these 3D culture systems with microfabricated platforms will be a powerful combination in compound screening and in the understanding of concentration-dependent biological effects while reducing cost and speeding discovery. Microfabricated systems offer the opportunity to grow cells under carefully controlled environmental conditions, for instance with gradients of growth and differentiation factors. A burgeoning number of microfluidic devices have been described for studying stem-cell renewal and differentiation, creating 3D spheroids, and drug testing.17–29 Most devices have utilized tumor cells grown in one or multiple chambers to mimic a limited aspect of gut function, such as absorption.30–33 The development of a microfluidic device compatible with the 3D culture of primary colonic epithelium remains a critical need for growth factor screening especially that involved in stem-cell renewal.
This report focuses on the adaptation, characterization, and implementation of a simple gradient-generating microfluidic device to assess the required concentration of Wnt-signaling factors on murine stem/transit-amplifying cell renewal and differentiation, and viability of primary colonic epithelial tissue in the colonoid system. A mouse model system was used since a wide range of disease models are available in mice. Additionally transgenic mice with EGFP and DsRED reporter systems are available to track expression of different proteins. Colonic crypts were loaded into the microdevice and developing colonoids were exposed to varying concentrations of the Wnt-pathway agonists, Wnt-3a and R-spondin1. The colonoids were characterized in situ over time by monitoring endogenously expressed fluorescent proteins and by immunochemistry to identify the presence of proliferative and differentiated cell types. The impact of the growth factors at varying concentrations and at varying culture times was quantified for large numbers of colonoids to provide statistically relevant data while minimizing reagent usage.
Experimental section
Transgenic mouse models and isolation of colonic crypts
Crypts were isolated from either Sox9EGFP mice or Sox9EGFP-CAGDsRed mice (6–9 weeks old) using previously described methods. The Sox9EGFP and Sox9EGFP-CAGDsRed mouse models were developed on a CD-1 background. The CAGDsRed mouse line ubiquitously expresses the red fluorescent protein DsRed under the control of a chicken beta-actin promoter (CAG). To create the Sox9EGFP-CAGDsRed mice, the DsRed-expressing mice were bred with Sox9EGFP mice, which possessed the Sox9 promoter controlling EGFP (enhanced green fluorescent protein) expression on a modified bacterial artificial chromosome.9,34 Mice genetically engineered with this construct express EGFP in intestinal stem cells and transit-amplifying cells. The colonic tissues were harvested from mice that were bred, handled and sacrificed under protocols approved by the UNC Institutional Animal Use and Care Committee.Colonoid culture
Colonoid culture media, CCM, consisted of a mixture of advanced DMEM/F12 medium (Life Technologies), Wnt-3A (120 ng mL−1) and R-spondin1 (175 ng mL−1) unless otherwise specified (Table S13†). CCM also contained noggin (100 ng mL−1), EGF (50 ng mL−1), Y27632 ROCK inhibitor (10 μM), NAC (1 mM), GlutaMAX (1×), HEPES (10 mM), penicillin (100 unit per mL), and streptomycin (100 μg mL−1). Wnt-3A and R-spondin1 were prepared from conditioned medium as described previously or purchased purified from a supplier (Table S13†).35 The CCM was prepared in a bulk volume of 500 mL, split into 6 mL aliquots, and stored at −80 °C until use. For crypt/colonoid culture, Matrigel was diluted 50% in CCM. A 1 mL suspension of freshly isolated crypts (5000 crypts per mL) was added to standard 12-well plates at 4 °C. The Matrigel was then polymerized for 15 min at 37 °C. After polymerization, 1.6 mL of CCM was overlaid onto the Matrigel. The isolated crypts typically formed colonoids within 24 h under these culture conditions. The CCM was changed every 24 h during the course of the experiment.Placement and culture of crypts and Matrigel on the gradient device
Before use, the device was sterilized with 70% ethanol and rinsed with phosphate buffered saline (PBS) ×5. The gradient-generating region of the device was coated by overnight incubation with 3% Matrigel in PBS for 12 h at 4 °C, and then rinsed with PBS ×3 prior to loading crypts or colonoids. This step resulted in deposition of a thick coat (35 ± 5 μm, n = 3) of Matrigel (Dow Corning, Midland, MI) on the channel walls (Fig. S2†) that improved adhesion of the subsequently loaded Matrigel plug, improved loading of the crypt/Matrigel suspension (see below) and centered subsequently loaded crypts/colonoids along the z axis of the device. Crypts were isolated from the distal colon of a mouse as previously described. The crypts were pelleted by centrifugation at 300 × g for 90 s. The supernatant was aspirated and the crypts were mixed with cold liquid Matrigel (50% in CCM, 4 °C). A 25 μL aliquot of this suspension containing 100 ± 10 crypts was pipetted into the device's gradient-generating region. The Matrigel pre-coat layer enabled the crypt/Matrigel solution to quickly enter the central channel by surface tension. Excess gel entering the reservoirs was removed and the gel was polymerized by incubation at 37 °C for 15 min. Once the Matrigel solidified, CCM (500 μL) was immediately added to each reservoir. For experiments in which a gradient was formed, Wnt-3A and/or R-spondin1 were omitted from the CCM added to the sink as appropriate for the specific experiment.Diffusion-based gradient generation and characterization
Gradient formation through the Matrigel layer on the device was characterized by imaging the movement of a 40 kDa FITC-dextran (Sigma-Aldrich, St. Louis, MO) in 50% Matrigel by time-lapse imaging using an Olympus MVX10 Macroview microscope. Fluorescence images were acquired every 15 min over 24 h to measure gradient formation. The volume of the source and sink was 500 μL and that of the channel was 5 μL. Gradient formation over time was modeled using Fick's Law:36where A is an integration constant, x ranges from 0 to 5 mm corresponding to the positions along the length of the channel, t is time, D is the diffusion coefficient, erfc is the complementary error function, and CO is the concentration of the species of interest loaded into the source. COMSOL Multiphysics with finite-element analysis (FEA) was used to model the data and calculate D. For experiments applying gradients to colonoids, the media in both the source and sink were replaced every 24 h.

Microscopy
Colonoid formation and growth over time was tracked by wide-field imaging of the entire device using an Olympus MVX10 research macro zoom fluorescence microscope with a 1.0×, 0.25 N.A. objective and 0.63× demagnification that provided a depth-of-focus of 91 μm. The MVX-10 was equipped with Chroma 49002 FITC/Cy2 and Chroma 49008 mCherry/Texas Red filter sets. Digital images were collected with a Hamamatsu Orca-flash 4.0 CCD camera. Confocal images of isolated crypts and colonoids were obtained using a Zeiss CLSM 710 Spectral Laser Scanning Microscope equipped with 405, 488 and 543 nm lasers to image Hoechst 33342, EGFP and DsRed, respectively. A Nikon Eclipse TE2000 microscope fitted with a Photometrics CoolSNAP HQ2 digital camera was used to quantify colonoid buds.On-chip fluorescence staining
Crypts isolated from a Sox9EGFP-only mouse were used for immunofluorescence staining to avoid interference from the DsRed fluorescence of the CAG-DsRed/Sox9EGFP mouse. For immunofluorescence staining, freshly isolated crypts, and colonoids on the device and on tissue-culture plates were fixed with 4% paraformaldehyde for 20 min, followed by permeabilization with 0.5% Triton X-100 (Thermo-Fisher, Waltham, MA) for 20 min. Following rinsing ×3 with PBS containing 100 mM glycine, the colonoids were incubated in immunofluorescence wash (0.2% Triton X-100, 0.1% BSA, 0.05% Tween-20, 7.7 mM NaN3 in PBS and 5% normal goat serum) for 90 min to block nonspecific binding. Primary-antibodies (polyclonal rabbit α-Muc2 [1:200] and polyclonal goat α-chromogranin A [1:1000]) were applied in immunofluorescence wash for 12 h at 4 °C (Life Technologies, Carlsbad, CA). Secondary antibodies (α-rabbit-Cy3 or α-goat-Cy3) were applied in immunofluorescence wash (1:500) for 45 min (Life Technologies, Carlsbad, CA). All nuclei were stained with Hoechst 33342 (10 μg mL−1 in PBS) using a 30 min incubation. Microdevices were imaged by brightfield and fluorescence microscopy. An EdU-based assay was also used to measure proliferating cells (Life Technologies, product #10640).On-chip quantification of colonoid fluorescence and area
For imaging colonoid formation under each of the gradient conditions, fluorescence images were acquired every 24 h for a total of 5 days. Microscopy with a large depth-of-focus of 91 μm was used so that the majority of the colonoid volume resided within the image plane. A custom script was written in MATLAB (MathWorks; Natick, MA) to quantify the number of DsRed-positive pixels and the EGFP fluorescence intensity of pixels in each colonoid in the 2D image. Prior to quantitation, images from the DsRed channel were pre-processed to reduce background noise using top-hat filtering.37 The images were then thresholded using Otsu's method38 and “holes” were closed to identify the number of pixels occupied by each colonoid in the device. The number of pixels was then converted to the area occupied by the colonoid in the 2-D image. When manually reexamined, this strategy yielded zero false negatives (missed colonoids) and 2% false positives (structures misidentified as a colonoid) for n = 1050 colonoids. In addition to identifying the colonoids, the number of DsRed-positive pixels or colonoid area was also used as a proxy for colonoid size or total cell number. The images from the EGFP channel were pre-processed to reduce background noise (top-hat filtering) and the fluorescence intensity of each pixel previously identified as being within the boundaries of colonoid (using the DsRed mask) was summed. The MATLAB script also included code to bin data from each of the devices according to the location on the device. For data analysis, the images of the gradient channel were divided into 4 regions each corresponding to a 1.25 mm length of the channel. The region adjacent to the source was always designated “region 1” while that nearest the sink was designated “region 4”.Boxplots were used to represent the non-normal distribution of colonoid area and EGFP fluorescence intensity of the developing colonoids.39 Within the boxplots, stars represented the mean, a bar represented the median, and the upper and lower boxes showed the 75% and 25% percentile of the data, respectively. The whiskers extended to the 5th and 95th percentile with outlying data shown as individual points. The data are presented in the text as medians, first- and third-quartile values for colonoid DsRed area and colonoid EGFP fluorescence intensity within the regions. For statistical comparison, the data were converted to a normal distribution using a logarithmic transform and then assessed using Q–Q plots for their fit to a Normal distribution. The adjusted coefficient of determination (R2) values for the Q–Q plots was always ≥0.93 (Fig. S7 and Table S15†). Statistical differences between data were identified using a Holm-Sidak t test in the analysis of variance.40 Data are also presented as average ± standard deviation where appropriate.
Off-chip quantification of colonoids possessing different fluorescent signatures
Crypts isolated from wild-type, Sox9EGFP mice were cultured in CCM at the indicated Wnt-3A and R-spondin1 concentrations in 12-well plates. After 5 days in culture, colonoids were fixed, and stained with a fluorescent marker as described above. Hoechst 33442 staining was used to identify nuclei. Imaging was performed at low resolution (91 μm depth of field) so that the entire colonoid was captured in a single image plane. Blue Hoechst fluorescence was used to identify and segment colonoids. All other image processing was as described above. Based on percentages obtained when freshly isolated crypts were stained and assayed, a colonoid was judged to possess goblet cells if the number of pixels positive for Muc-2 was greater than 10% of the total colonoid pixel number. A colonoid was considered to possess enteroendocrine cells if the pixels positive for Chg-A was greater than 0.5% the total colonoid pixel number. All data sets reflect n ≥ 20 colonoids.Off-chip quantification of the colonoid volume displaying a fluorescent signature
Crypts isolated from wild-type, Sox9EGFP mice were cultured in CCM at the indicated Wnt-3A and R-spondin1 concentrations in 12-well plates. After 5 days in culture, colonoids were fixed, and stained with a fluorescent marker (immunofluorescence, EdU or other) as described above. Hoechst 33442 staining was used to identify nuclei and segment the colonoids in three dimensions. The colonoids were imaged confocally to obtain a set of image slices covering the entire volume of the colonoid. Thresholding and masking were performed for each confocal slice as described in the prior section. To quantify the percentage of pixels in a colonoid possessing a fluorescent marker, the number of pixels positive for the fluorescent marker in every image slice of that colonoid was divided by the total number of pixels in the colonoid. This was then reported as the percentage of colonoid volume positive for the fluorescent marker. All data sets reflect at least n = 5 colonoids.Results and discussion
Gradient characterization
The current work focused on the adaptation, characterization and utilization of a simple, gradient-generating microdevice to assess the dose-dependent effects of the two principle Wnt-signaling proteins, Wnt-3a and R-spondin1, on colonic stem/transit-amplifying-cell activity using the colonoid as an in vitro model system. PDMS was selected as the material of choice for the device since PDMS microdevices can be readily prepared on a benchtop, are gas permeable, and are compatible with colonic stem cells.41 The device design was simple, incorporating a central 5 mm-long microchannel with a large reservoir at either end. The gradient was formed across the microchannel which was filled with Matrigel (Fig. S2†). The reservoir volumes were 100× that of the gradient forming region.42,43FITC-dextran (40 kDa) was used as a model analyte to characterize the gradient formed on the device as it is similar in molecular weight to Wnt-3a (39.7 kDa) and R-spondin1 (40.0 kDa). The microchannel was imaged over time by fluorescence microscopy after addition of a solution of the fluorescent dextran to the source reservoir. At times after 24 h, the measured fluorescence through the microchannel displayed a linear decrease from the source to the sink reservoirs (Fig. 1B). The temporal evolution of the fluorescence intensity across the microchannel was fit to Fick's Law. The experimentally measured molecular diffusion coefficient of the FITC-dextran was 7.4 ± 0.5 × 10−11 m2 s−1, which is similar to that measured for vascular epithelial growth factor (42 kDa) through Matrigel (7.0 × 10−11 m2 s−1).44,45 To maintain this linear gradient over long time scales, the source and sink solutions were replaced every 24 h. Construction of a model incorporating these solution changes indicated that the concentration of a 40 kDa analyte across the microchannel will vary by no more than 0.3% over a 5 day period. These data indicated that a stable, linear gradient was successfully established across the Matrigel plug within the microchannel between the source and sink reservoirs. Similar gradient strategies have been employed successfully by others.42,43
 | ||
| Fig. 1 Characterization of the gradient-generating microdevice. (A) Photograph of the device. The Matrigel-filled gradient region resides between the source (left with yellow dye) and sink (right with blue dye) reservoirs. (B) Gradient characterization on the microdevice. Movement of a 40 kDa FITC-dextran through the channel was monitored using time-lapse fluorescence imaging. The experimentally measured data is marked as red stars. The solid black line is the fit to the data using Fick's Law. (C and D) Freshly isolated crypts and colonoids (Sox9EGFP-only mice) cultured for 5 days on the microdevice were stained for goblet cells (Muc2) (C), and enteroendocrine cells (ChgA) (D) and imaged by confocal microscopy. Nuclei were stained with Hoechst 33342 (blue). (E) Crypts/colonoids treated as in panels (C and D) but imaged for EGFP (EGFP-Sox9) which marks stem/transit amplifying cells (E). (F) Histogram showing percentages of colonoids possessing goblet cells (Muc2+), enteroendocrine cells (ChgA+), and stem/transit-amplifying cells (EGFP-Sox9+) in colonoids cultured on the microdevice (white bars) or conventional Matrigel-patty culture (grey bars). | ||
Comparison of colonoids cultured on the microdevice to that cultured under standard conditions
To determine whether culture within the PDMS device altered colonoid formation and growth, freshly isolated crypts were mixed with Matrigel and loaded into the microchannel. CCM containing typical concentrations of both Wnt-3a (120 ng mL−1) and R-spondin1 (175 ng mL−1) for colonoid culture was placed into both the source and sink reservoirs of the microdevice and replenished every 24 h during culture. In parallel, crypts were cultured in a conventional Matrigel patty overlaid with the identical CCM which was also replaced every 24 h. Crypts from a Sox9EGFP-CAGDsRed mouse were used since the expression of DsRed in all cells and EGFP in stem/transit-amplifying cells enabled rapid assessment of both colonoid size (DsRed+) and the relative number of proliferative cells (EGFP+).9 Of the crypts plated in the microdevice, 62.0 ± 12.5% (avg. ± s.d.) developed into colonoids with a median DsRed area of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 010 μm2 after 5 days in culture (Table S2†). In comparison, 63.5 ± 7.5% of crypts plated and cultured for 5 days in the Matrigel patties under standard conditions developed into colonoids with a median DsRed area of 16240 μm2 (Table S2†). The percentage of colonoids possessing EGFP expression was similar under both conditions with 82.0 ± 7.0% (microdevice) and 80.5 ± 6.0% (control) of colonoids positive for EGFP. The average EGFP fluorescence intensity per colonoid on the entire device increased from day 1 to day 5. The median integrated EGFP fluorescence per colonoid on day 1 and day 5 was 23250 and 62830 RFUs, respectively, suggesting that the colonoids possessed actively dividing populations of colonic stem cells (Table S3†). In comparison, colonoids cultured for 5 days in the Matrigel patties under standard conditions developed into colonoids with an average EGFP fluorescence similar to that of colonoids cultured on the microdevice. The median EGFP fluorescence per colonoid on day 1 and day 5 was 26810 and 66610 RFUs, respectively (Table S3†). These data demonstrated that the rate of colonoid formation and growth and the numbers of stem/transit-amplifying cells increased within expanding colonoids in the microdevice in a manner similar to that in conventional Matrigel patties.
010 μm2 after 5 days in culture (Table S2†). In comparison, 63.5 ± 7.5% of crypts plated and cultured for 5 days in the Matrigel patties under standard conditions developed into colonoids with a median DsRed area of 16240 μm2 (Table S2†). The percentage of colonoids possessing EGFP expression was similar under both conditions with 82.0 ± 7.0% (microdevice) and 80.5 ± 6.0% (control) of colonoids positive for EGFP. The average EGFP fluorescence intensity per colonoid on the entire device increased from day 1 to day 5. The median integrated EGFP fluorescence per colonoid on day 1 and day 5 was 23250 and 62830 RFUs, respectively, suggesting that the colonoids possessed actively dividing populations of colonic stem cells (Table S3†). In comparison, colonoids cultured for 5 days in the Matrigel patties under standard conditions developed into colonoids with an average EGFP fluorescence similar to that of colonoids cultured on the microdevice. The median EGFP fluorescence per colonoid on day 1 and day 5 was 26810 and 66610 RFUs, respectively (Table S3†). These data demonstrated that the rate of colonoid formation and growth and the numbers of stem/transit-amplifying cells increased within expanding colonoids in the microdevice in a manner similar to that in conventional Matrigel patties.
The presence of differentiated cell lineages, goblet and enteroendocrine cells, in colonoids on the microdevice was also compared to that of colonoids under conventional culture conditions (Fig. 1A–C). At 5 days after plating freshly isolated crypts, the colonoids in the microchannel were assayed for these lineages and compared with controls cultured under standard conditions. The percentage of colonoids on the microdevice expressing goblet cells (Muc-2+) was 95.0 ± 3.5%, compared to 92.5 ± 5.5% in the Matrigel patty (Fig. 1D). The percentage of colonoids containing enteroendocrine cells (ChgA+) was 38.5 ± 10.0% on the microdevice, which was similar to that for colonoids in standard culture (34.5 ± 13.5%) (Fig. 1D). These data demonstrated that colonoids cultured on the microdevice and in the Matrigel patty developed similarly in terms of the presence of differentiated cell types.
Comparison of colonoids cultured in different regions of the microchannel in the absence of a gradient
Due to the length of the microchannel, it was important to determine whether colonoids developed identically throughout the length of the channel in the absence of a gradient. Crypts from a Sox9EGFP-CAGDsRed mouse were loaded and cultured in Matrigel on the microdevice with CCM containing Wnt-3a (120 ng mL−1) and R-spondin1 (175 ng mL−1) in both the source and sink reservoirs. The properties of the colonoids in each of the 4 regions of the channel were quantified from images acquired daily over 5 days (Fig. 2A). To compare colonoid size in each region, the DsRed area per colonoid was determined for each of the 4 regions. Ten separate devices were assayed (n = 253, 277, 266, 254 total number of colonoids after 5 days in regions 1–4, respectively). Colonoids expanded in all regions of the microdevice and the median DsRed area per colonoid was 15731 μm2 (region 1), 12767 μm2 (region 2), 13930 μm2 (region 3) and 13320 μm2 (region 4) (Fig. 2C and D and Table S4†). To assess stem/transit-amplifying cell renewal and expansion in each of the four regions across the device, EGFP fluorescence was measured. At day 5, the median EGFP fluorescence per colonoid was 56157 RFUs (region 1), 66039 RFUs (region 2), 58758 RFUs (region 3) and 58766 RFUs (region 4) (Fig. 2E and F and Table S5†). When the different regions were compared, the differences in the DsRed area per colonoid and EGFP fluorescence were not statistically different. Based on these data, colonoid growth and stem/transit-amplifying cell number was similar in all regions of the microchannel.
 | ||
| Fig. 2 Culture of colonoids in the absence of a gradient. Colonoid data is shown at days 1 (A, C and E) and 5 (B, D and F) of culture. Overlaid DsRed-EGFP fluorescence images of the colonoids are shown (A and B). The scale bar represents 500 μm. Boxplots were used to represent the non-normal distribution of the area (C and D) or EGFP-Sox9 fluorescence (E and F) per colonoid. Colonoid area is represented as μm2 (×104) and integrated EGFP fluorescent intensity is represented as RFUs (×105). For the boxplots, the red stars indicate the mean of the data, the bar shows the median, and the upper and lower boxes represent the 75% and 25% of the data, respectively. The whiskers extend to the 5% and 95% with the individual points showing outliers. | ||
Effect of Wnt-3a concentration on colonoid expansion and stem/transit-amplifying cell number
Crypts were loaded into the microchannel as above, but Wnt-3a (120 ng mL−1) was placed in the source reservoir while medium lacking Wnt-3a was placed in the sink reservoir. The R-spondin1 concentration was held constant at 175 ng mL−1 in both reservoirs. Seven separate devices were assayed to examine the effect of the Wnt-3a concentration on the colonoids (Table S1†). After 24 h in culture, the differences in the DsRed area per colonoid were not statistically significant across the four device regions suggesting that at this early time the size of colonoids developing from the crypts was similar at all Wnt-3A concentrations (Fig. S3A and C†). However, after 5 days in culture, the average DsRed area per colonoid (n = 181, 173, 194, 179 colonoids in regions 1–4, respectively) varied considerably between regions (Fig. 3B and D and Table S6†). The largest colonoids were present in regions 1 and 2 ([Wnt-3a] > 60 ng mL−1) with median areas of 10721 μm2 and 8960 μm2, respectively. The area of colonoids cultured in regions 3 and 4 ([Wnt-3a] < 60 ng mL−1) demonstrated median areas of 8566 and 4610 μm2, respectively. A comparison of colonoid size in all of the different lanes demonstrated that region 4 was statistically different from regions 1 and 2 (p < 0.05) and region 3 was statistically different from region 1 (p < 0.05). The impact of Wnt-3a concentration on the stem/transit amplifying cell number within the colonoids was also assessed. After 5 days in culture, the colonoids in regions 1 and 2 possessed much greater EGFP fluorescence per colonoid than those in regions 3 and 4 with median EGFP fluorescence of 68773, 39738, 20605 and 12082 RFUs, for regions 1–4, respectively (Fig. S3 and Table S7†). Comparison of eGFP fluorescence (stem/transit amplifying cell number) in all of the different lanes demonstrated that regions 3 and 4 were statistically different from regions 1 and 2 (p < 0.05). These data revealed a dose-dependent impact on stem/transit-amplifying cell proliferation in response to [Wnt-3a], consistent with a minimal required concentration of 60 ng mL−1 i.e. the concentration at the boundary between regions 3 and 2. This concentration is well below that used for colonoid culture in the vast majority of publications (250–500 ng mL−1).1,9 Thus, current accepted in vitro culture conditions appear to be utilizing a vast excess of Wnt-3a well above the threshold needed for stem-cell renewal and maintenance.
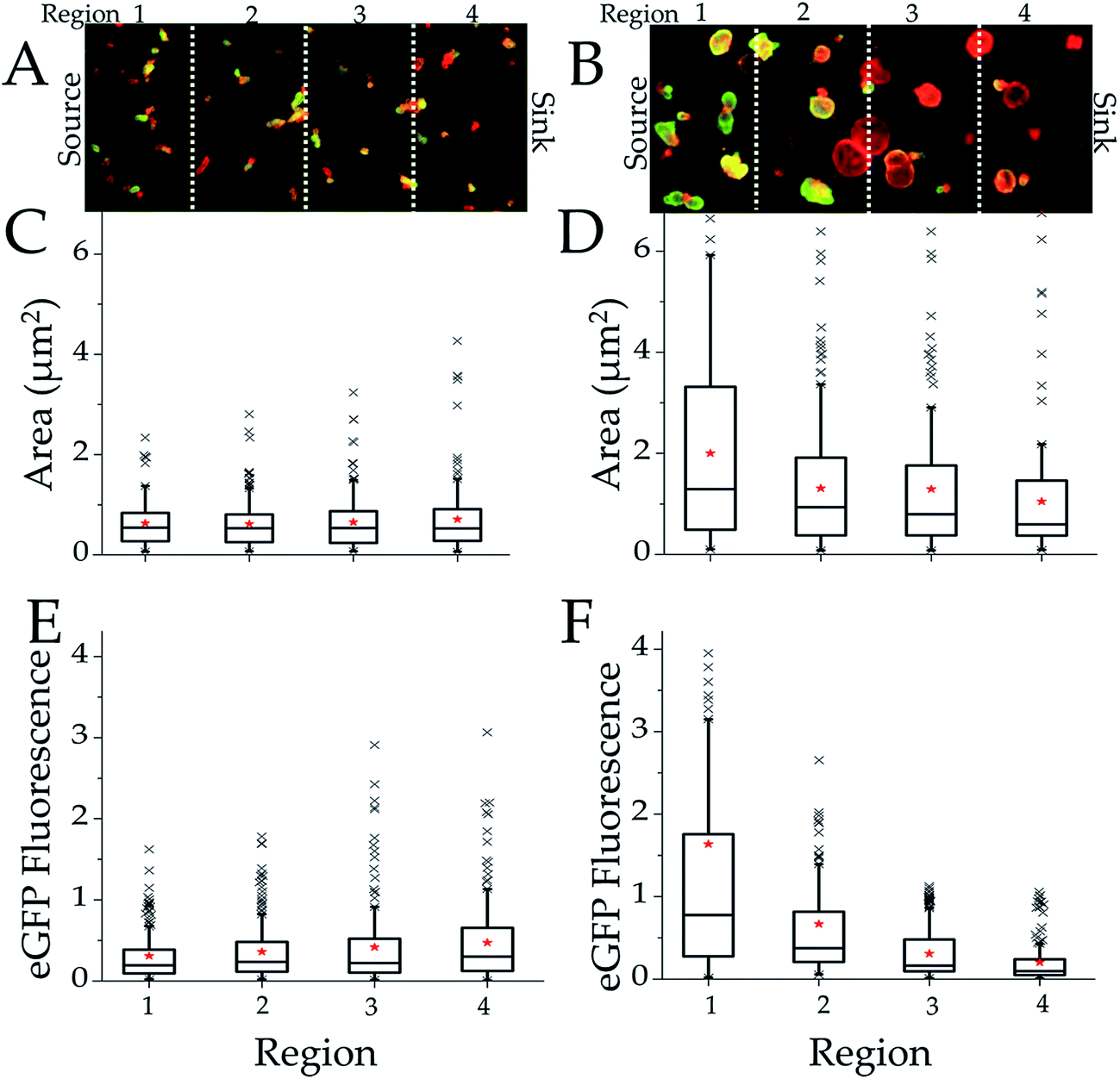 | ||
| Fig. 3 Culture of colonoids in the presence of a combined R-spondin1 and Wnt-3a gradient. Colonoid data is shown at days 1 (A, C and E) and 5 (B, D and F) of culture. Overlaid DsRed-EGFP fluorescence images of the colonoids are shown (A and B). The scale bar represents 500 μm. Boxplots as described in the legend of Fig. 2 were used to represent the non-normal distribution of the area (C and D) or EGFP-Sox9 fluorescence (E and F) per colonoid. Colonoid area is represented as μm2 (×104) and integrated EGFP fluorescent intensity is represented as RFUs (×105). The Wnt-3a and R-spondin1 concentrations in the sink were 0 ng mL−1 while that in the source was and 120 and 175 ng mL−1, respectively. The threshold concentration of each factor occurred at the interface between regions 2 and 3 and is marked by the yellow arrow. | ||
Effect of R-spondin1 concentration on colonoid expansion and stem/transit-amplifying cell number
Crypts were cultured in the microchannel under a linear R-spondin1 gradient (0–175 ng mL−1). At 24 h, the DsRed area per colonoid was similar across the 4 regions of the microchannel (Fig. S4A and C†). After 5 days in culture, the largest colonoids developed in regions 1–3 at [R-spondin1] > 44 ng mL−1 with median DsRed areas per colonoid of 9870, 11798, 8857, and 5569 μm2, for regions 1–4, respectively (Fig. S4B and D and Table S8†). However when all regions were compared, only regions 4 and 1 were statistically different (p < 0.05) with respect to DsRed fluorescence. Six separate devices were assayed for these data (n = 162, 152, 181, 164 total number of colonoids after 5 days in regions 1–4, respectively). The impact of R-spondin1 concentration on stem/transit-amplifying cell numbers was also assessed by measuring colonoid EGFP fluorescence. After 5 days in culture, colonoids in regions 1 and 2 possessed statistically significant greater levels of EGFP fluorescence per colonoid relative to region 4 (p < 0.05) while region 3 was significantly different from region 2 (p < 0.05). Median EGFP fluorescence was 54298, 59967, 34149 and 43982 RFUs, respectively in regions 1–4 (Fig. S4B and F and Table S9†). Taken together, the data on colonoid area and stem/transit-amplifying cell proliferation support a minimal concentration for the bioactivity of R-spondin1 near 88 ng mL−1 i.e. that concentration occurring near the interface of regions 2 and 3. This R-spondin1 concentration is well below that which has been empirically used in colonoid culture systems (500–1000 ng mL−1),1,9 again suggesting that current in vitro culture conditions utilize an R-spondin1 concentration well above that needed to support stem-cell renewal.
Colonoid growth in the presence of combined Wnt-3a and R-spondin1 gradients
In the prior experiments, the concentration of one of the growth factors was held constant at a supra-threshold concentration while the other was varied. To understand whether colonoids could expand and maintain stem cells when cultured in the presence of both Wnt-3a and R-spondin1 at lower concentrations, colonoids were cultured on the gradient device under conditions in which the minimal required concentrations of Wnt-3a and R-spondin1 coincided at the interface of regions 2 and 3. Wnt-3a (120 ng mL−1) and R-spondin1 (175 ng mL−1) were added to the source reservoir, but were excluded from the sink (Table S1†). Six separate devices were assayed (n = 147, 163, 152, 148 total number of colonoids after 5 days in regions 1–4, respectively). At 24 h of culture, the 4 channel regions possessed similar DsRed area per colonoid (Fig. 3A and C, and Table S10†). After 5 days in culture, colonoids in region 1 experienced a >3-fold increase in area with the median value of 13923 μm2 (Fig. 3 and Table S10†). Over this same timescale, colonoids cultured in regions 2, 3, and 4 increased their median areas by 1.8, 1.5 and 1.1-fold, respectively. The differences between colonoid sizes in regions 1–3, 1–4, and 2–4 were statistically significant (p < 0.05). When the EGFP fluorescence was examined, the colonoids in regions 1 and 2 increased their median integrated EGFP fluorescence intensity by 4.0- and 1.6-fold, respectively between days 1 and 5. Regions 3 and 4 on the other hand displayed an overall decrease in EGFP fluorescence, with a 0.7- and 0.3-fold decrease in EGFP fluorescence per colonoid, respectively, between days 1 and 5 (Table S11†). The differences in EGFP fluorescence per colonoid in each of regions 1–2 was statistically different from that in each of lanes 3 and 4 (p < 0.05). Taken together these data suggested that the required minimal concentrations of Wnt-3a (>60 ng mL−1) and R-spondin1 (>88 ng mL−1) were independent of each other and both factors were required to support proliferation and growth of the stem/transit amplifying cells in the colonoids.
Comparison of growth factor reduced to conventional culture conditions
In the current literature, a wide range of growth factor concentrations are employed for colonoid maintenance during experimentation, with the majority of groups utilizing a [Wnt-3a] > 100 ng mL−1 (ref. 13) and R-spondin1 > 1000 ng mL−1.1,9,10,14,34,35 In contrast, the gradient-device data suggests that significantly lower concentrations of these factors [Wnt-3a (60 ng mL−1) and R-spondin1 (88 ng mL−1)] will maintain stem cells and sustain colonoid cultures. For this reason, these growth-factor concentrations were compared to those used in conventional culture to determine whether colonoid attributes were similar under the two conditions. Sox9EGFP-CAGDsRed crypts were cultured within a Matrigel patty in 12-well plates and six parameters were compared: (i) growth based on change in colonoid area over time, (ii) differentiation based on presence of goblet and enteroendocrine cells, (iii) stem cell maintenance/support based on renewal of stem/transit amplifying cells, (iv) cell proliferation based on EdU staining, (v) maintenance in culture based on passaging efficiency, and (vi) morphologic characteristics based on colonoid bud formation.At 24 h in culture, the average colonoid area was 6271 ± 852 and 6818 ± 930 μm2 for growth factor reduced and conventional conditions, respectively.9 After 5 days in culture, colonoids continued to maintain similar areas, 28129 ± 2309 and 29621 ± 2957 μm2 under both conditions (Fig. 4A). The presence of goblet cells (Muc2), enteroendocrine cells (ChgA), stem/transit amplifying cells (EGFP-Sox9) and actively proliferating cells (EdU-based assay) in the colonoids were also similar at day 5 (Fig. 4C and S6†). A greater percentage of the colonoid volume was occupied by enteroendocrine cells (ChgA+) than that of colonoids under conventional culture (1.2 ± 0.2% versus 0.6 ± 0.2%, p ≤ 0.05). The numbers of these rare cells were still low compared to normal crypts which possess ∼6% ChgA+ cells.43 When all assays were considered, the two culture systems yielded similar numbers of stem/transit amplifying and differentiated cells.
 | ||
| Fig. 4 Properties of colonies cultured under growth factor reduced (Wnt-3a: 60 ng mL−1, R-spondin1: 88 ng mL−1) vs. conventional (Wnt-3a: 100 ng mL−1, R-spondin1: 1000 ng mL−1) conditions. (A) Colonoid size at day 1 (light gray) and 5 (dark gray) of culture. (B) Representative image of colonoids cultured under the conventional conditions and the reduced growth factor conditions. Scale bar is 200 μm. (C) Upper two panels: immunofluorescence staining of mucin 2 (Muc2) and chromogranin A (ChgA) for colonoids grown under conventional (light grey) and growth reduced growth factor (dark grey) conditions. Lower left panel: EdU-based assay for proliferation. Lower right panel: eGFP expressed under a Sox9 promoter. Each data set represents n = 5 colonoids. (D) Colonoid solidity plotted against colonoid area divided by colonoid perimeter. Each symbol represents a single colonoid cultured under either the conventional (square) or reduced growth factor (circle) conditions. Solid symbols (square or circle) represent the colonoids that were classified as budding and open symbols (square or circle) represent the colonoids that are classified as non-budding. (E) Quantification of the number of colonoids formed after 3 passages (P1, P2, P3) in conventional (light grey) growth reduced growth factor (dark grey) conditions. | ||
To compare the morphological characteristics of the colonoids grown under both conditions, the presence of buds or multi-cellular protrusions around the central lumen of individual colonoids was assessed. These buds house collections of stem cells and previous work by Sato and colleagues suggests that budding might be an early stage of crypt formation.14 Thus colonoids with greater budding profiles are likely to be more representative of a normal phenotype. Two colonoid attributes (solidity and area divided by perimeter) were utilized as a metric for the presence of bud formation around a central lumen. The solidity defined as the colonoid area divided by the convex hull area in the 2D image measured the extent to which the colonoid area was studded with concave cavities such as might occur between buds. Whereas the area divided by perimeter is more reflective of how convoluted the colonoid surface is. A training data set of manually identified budding and nonbudding colonoids combined with support vector machine learning was used to classify test colonoids grown under the different culture conditions as either budding or non-budding (Fig. S5†). The majority of colonoids cultured under the reduced growth factor conditions (92 ± 6%, n = 3 experimental replicates of 25 colonoids each) were classified as possessing buds, whereas only 8 ± 4% of colonoids (n = 3 experimental replicates of 25 colonoids each) cultured under the conventional conditions were scored as possessing buds (Fig. 4D). The much greater number of colonoids possessing buds under the reduced factor conditions suggested that these conditions promoted more appropriate gut morphologic patterning than the higher concentrations of Wnt-3a and R-spondin1.11,46 These data are also consistent with the greater number of ChgA+ cells in the reduced-factor conditions observed previously. Colonoids cultured under the conventional factor conditions also displayed a more cystic morphology with thin outer walls relative to that under conventional conditions (Fig. 4B and S8†). Gracz and colleagues recently characterized the genotypic differences between the cystic and noncystic colonoid morphologies.47 While both phenotypes possess a central lumen, the noncystic phenotype displays a greater mRNA expression of proteins characteristic of differentiated lineages, whereas the cystic structures exhibit gene expression patterns consistent with high levels of Wnt signaling and cell turnover, but low levels of differentiation. Taken together these data suggest that the reduced-factor conditions promote a more morphologically relevant colonoid with a phenotype more similar to a crypt compared to the conventional culture conditions.
A critical attribute of any culture system is the efficiency of passage or the length of time that the culture system can be maintained in vitro. To assess the ability to grow colonoids under the reduced growth factor concentrations on long time scales, cells from freshly isolated crypts were plated at identical densities and cultured. After 5 days, the total number of colonoids arising from the plated cells were counted and then harvested, fragmented and 104 cells from this harvest re-plated in culture. This process was repeated every 5 days, for a total of 3 passages. Colonoid outgrowth was similar for both the reduced factor and conventional culture systems, with no statistical difference in the number of colonoids generated after each passage step (Fig. 4E). These data suggest that the identified, minimal concentrations of Wnt-3a and R-spondin1 did not affect bulk size or longevity of colonoid culture, but did produce a more morphologically appropriate mini-gut compared to standard culture conditions. Additionally, the reduction in factor concentrations needed to maintain the colonoids in culture is expected to lower the reagent cost of colonoid culture by 66% (Table S12†).
Conclusion
We describe the implementation of a microengineered technology to create tightly controlled linear gradients of morphogenic factors along a defined culture region housing a population of primary colonic organoids to enable efficient and rapid screening of cell proliferation and differentiation within the colonoids. The microdevice enabled a substantial reduction in the quantity of Matrigel and expensive growth factors needed to assay a wide range of factor concentrations for colonoid growth since the volume of the microchannel (10 μL) was small compared that on a 96-well plate. For example, 10 microwells of a standard 96-well plate would consume 1 mL of these reagents, 100× greater volume. The reduction in the assay volume needed to survey a wide range of factor concentrations would similarly greatly decrease the numbers of mice needing to be sacrificed to optimize factor concentrations. The decreased need for tissue would in turn translate to smaller breeding numbers and transgenic mouse colony size.The technology made possible the efficient elucidation of optimum protein factor concentrations for stem-cell renewal (proliferation) and colonoid growth. Colonoids were exposed to four experimental conditions: no gradient in a high Wnt-3a and high R-spondin1 environment, a Wnt-3a gradient in a high R-spondin1 environment, an R-spondin1 gradient in a high Wnt-3a environment, and a combined Wnt-3a and R-spondin1 gradient. Thresholds of 60 ng mL−1 of Wnt-3a and 88 ng mL−1 of R-spondin1 were the minimal concentrations of these factors required to stimulate stem cell proliferation and overall colonoid growth. Prior research utilizing cultured colonoids has in general used substantially greater concentrations with Wnt-3a concentrations up to 100 ng mL−1 and R-spondin1 concentrations up to 1000 ng mL−1.9,10 The overstimulation of Wnt signaling pathways in these colonoid culture systems may account for their paucity of absorptive enterocytes and excessive numbers of stem cells relative to that in normal colon. By utilizing the threshold concentrations of Wnt-3a and R-spondin1 identified in this work, a colonoid phenotype was generated displaying crypt-like budding and columnar morphology with greater expression of enteroendocrine lineages. Use of these reduced factor concentrations will permit more physiologically relevant colonoid culture conditions at significant cost savings by virtue of the reduced concentrations of expensive growth factors. The microfluidic device and protocols described in this series of experiments will enable intestinal biologists to pursue further in-depth combinatorial screens of factors and pharmacologic compounds for controlling colon stem-cell renewal and differentiation.
Acknowledgements
This research was supported by the NIH (F31CA183309, EY024556, and DK091427). We acknowledge the UNC-Olympus Imaging Research Center and the Microscopy Service Laboratory (MSL) for access to confocal microscopes.References
- T. Sato, R. G. Vries, H. J. Snippert, M. van de Wetering, N. Barker, D. E. Stange, J. H. van Es, A. Abo, P. Kujala, P. J. Peters and H. Clevers, Nature, 2009, 459, 262–265 CrossRef CAS PubMed
.
- L. G. van der Flier and H. Clevers, Annu. Rev. Physiol., 2009, 71, 241–260 CrossRef CAS PubMed
- X. C. He, J. W. Zhang, W. G. Tong, O. Tawfik, J. Ross, D. H. Scoville, Q. Tian, X. Zeng, X. He, L. M. Wiedemann, Y. Mishina and L. H. Li, Nat. Genet., 2004, 36, 1117–1121 CrossRef CAS PubMed
- A. Humphries and N. A. Wright, Nat. Rev. Cancer, 2008, 8, 415–424 CrossRef CAS PubMed
- T. Takayama, K. Miyanishi, T. Hayashi, T. Kukitsu, K. Takanashi, H. Ishiwatari, T. Kogawa, T. Abe and Y. Niitsu, Clin. Gastroenterol. Hepatol., 2005, 3, S42–S45 CrossRef
- M. K. Dame, Y. Jiang, H. D. Appelman, K. D. Copley, S. D. McClintock, M. N. Aslam, D. Attili, B. J. Elmunzer, D. E. Brenner, J. Varani and D. K. Turgeon, Lab. Invest., 2014, 94, 222–234 CrossRef CAS PubMed
- T. Grabinger, L. Luks, F. Kostadinova, C. Zimberlin, J. P. Medema, M. Leist and T. Brunner, Cell Death Dis., 2014, 5, e1228 CrossRef CAS PubMed
- A. Haegebarth and H. Clevers, Am. J. Pathol., 2009, 174, 715–721 CrossRef CAS PubMed
- S. Ramalingam, G. W. Daughtridge, M. J. Johnston, A. D. Gracz and S. T. Magness, Am. J. Physiol.: Gastrointest. Liver Physiol., 2012, 302, G10–G20 CrossRef CAS PubMed
- T. Sato, D. E. Stange, M. Ferrante, R. G. J. Vries, J. H. van Es, S. van den Brink, W. J. van Houdt, A. Pronk, J. van Gorp, P. D. Siersema and H. Clevers, Gastroenterology, 2011, 141, 1762–1772 CrossRef CAS PubMed
- M. Stelzner, M. Helmrath, J. C. Y. Dunn, S. J. Henning, C. W. Houchen, C. Kuo, J. Lynch, L. H. Li, S. T. Magness, M. G. Martin, M. H. Wong, J. Yu and N. I. H. I. S. C. Consortiu, Am. J. Physiol.: Gastrointest. Liver Physiol., 2012, 302, G1359–G1363 CrossRef CAS PubMed
- H. Clevers, Nature, 2013, 495, 53–54 CrossRef CAS
- A. Ootani, X. N. Li, E. Sangiorgi, Q. T. Ho, H. Ueno, S. Toda, H. Sugihara, K. Fujimoto, I. L. Weissman, M. R. Capecchi and C. J. Kuo, Nat. Med., 2009, 15, U1–U140 CrossRef PubMed
- T. Sato and H. Clevers, Science, 2013, 340, 1190–1194 CrossRef CAS PubMed
- S. Yui, T. Nakamura, T. Sato, Y. Nemoto, T. Mizutani, X. Zheng, S. Ichinose, T. Nagaishi, R. Okamoto, K. Tsuchiya, H. Clevers and M. Watanabe, Nat. Med., 2012, 18, 618–623 CrossRef CAS PubMed
- X. Yin, H. F. Farin, J. H. van Es, H. Clevers, R. Langer and J. M. Karp, Nat. Methods, 2014, 11, 106–112 CrossRef CAS PubMed
- B. G. Chung and J. Choo, Electrophoresis, 2010, 31, 3014–3027 CrossRef CAS PubMed
- M. Tehranirokh, A. Z. Kouzani, P. S. Francis and J. R. Kanwar, Biomicrofluidics, 2013, 7, 051502 CrossRef PubMed
- Y. S. Zhang, A. Sevilla, L. Q. Wan, I. R. Lemischka and G. Vunjak-Novakovic, Stem Cells, 2013, 31, 1806–1815 CrossRef CAS PubMed
- B. P. Mahadik, T. D. Wheeler, L. J. Skertich, P. J. A. Kenis and B. A. C. Harley, Adv. Healthcare Mater., 2014, 3, 449–458 CrossRef CAS PubMed
- G. H. Kwon, Y. Y. Choi, J. Y. Park, D. H. Woo, K. B. Lee, J. H. Kim and S.-H. Lee, Lab Chip, 2010, 10, 1604–1610 RSC
- C. Kim, S. Chung, Y. E. Kim, K. S. Lee, S. H. Lee, K. W. Oh and J. Y. Kang, Lab Chip, 2011, 11, 246–252 RSC
- S. Allazetta, T. C. Hausherr and M. P. Lutolf, Biomacromolecules, 2013, 14, 1122–1131 CrossRef CAS PubMed
- D. J. Alcendor, F. E. Block, D. E. Cliffel, J. S. Daniels, K. L. J. Ellacott, C. R. Goodwin, L. H. Hofmeister, D. Li, D. A. Markov, J. C. May, L. J. McCawley, B. McLaughlin, J. A. McLean, K. D. Niswender, V. Pensabene, K. T. Seale, S. D. Sherrod, H.-J. Sung, D. L. Tabb, D. J. Webb and J. P. Wikswo, Stem Cell Res. Ther., 2013, 4(suppl. 1), S18 CrossRef PubMed
- L. Evensen, D. R. Micklem, W. Link and J. B. Lorens, Cytometry, Part A, 2010, 77, 41–51 Search PubMed
- Y. Liu, D. A. Markov, J. P. Wikswo and L. J. McCawley, Biomed. Microdevices, 2011, 13, 837–846 CrossRef PubMed
- L. Y. Wu, D. Di Carlo and L. P. Lee, Biomed. Microdevices, 2008, 10, 197–202 CrossRef CAS PubMed
- L. Yu, M. C. W. Chen and K. C. Cheung, Lab Chip, 2010, 10, 2424–2432 RSC
- J. H. Sung and M. L. Shuler, Lab Chip, 2009, 9, 1385–1394 RSC
- H. Kimura, T. Yamamoto, H. Sakai, Y. Sakai and T. Fujii, Lab Chip, 2008, 8, 741–746 RSC
- J. H. Yeon and J.-K. Park, Anal. Chem., 2009, 81, 1944–1951 CrossRef CAS PubMed
- Y. Imura, Y. Asano, K. Sato and E. Yoshimura, Anal. Sci., 2009, 25, 1403–1407 CrossRef CAS
- P. M. van Midwoud, M. T. Merema, E. Verpoorte and G. M. M. Groothuis, Lab Chip, 2010, 10, 2778–2786 RSC
- A. D. Gracz, S. Ramalingam and S. T. Magness, Am. J. Physiol.: Gastrointest. Liver Physiol., 2010, 298, G590–G600 CrossRef CAS PubMed
- A. A. Ahmad, Y. Wang, A. D. Gracz, C. E. Sims, S. T. Magness and N. L. Allbritton, J. Biol. Eng., 2014, 8, 9 CrossRef PubMed
- J. Crank, The mathematics of diffusion, Oxford university press, 1979 Search PubMed
- X. Z. Bai, Appl. Opt., 2013, 52, 3777–3789 CrossRef PubMed
- H.-F. Ng, Pattern Recogn. Lett., 2006, 27, 1644–1649 CrossRef PubMed
- M. Frigge, D. C. Hoaglin and B. Iglewicz, Am. Stat., 1989, 43, 50–54 Search PubMed
- S. A. Glantz, Primer of Biostatistics, McGraw-Hill, 2005 Search PubMed
- Y. Wang, A. A. Ahmad, P. K. Shah, C. E. Sims, S. T. Magness and N. L. Allbritton, Lab Chip, 2013, 13, 4625–4634 RSC
- V. V. Abhyankar, M. W. Toepke, C. L. Cortesio, M. A. Lokuta, A. Huttenlocher and D. J. Beebe, Lab Chip, 2008, 8, 1507–1515 RSC
- V. V. Abhyankar, M. A. Lokuta, A. Huttenlocher and D. J. Beebe, Lab Chip, 2006, 6, 389–393 RSC
- R. Roskoski Jr, Crit. Rev. Oncol. Hematol., 2007, 62, 179–213 CrossRef PubMed
- R. R. Chen, E. A. Silva, W. W. Yuen and D. J. Mooney, Pharm. Res., 2007, 24, 258–264 CrossRef CAS
- N. Barker, S. Tan and H. Clevers, Development, 2013, 140, 2484–2494 CrossRef CAS PubMed
- A. D. Gracz, I. A. Williamson, K. C. Roche, M. J. Johnston, F. Wang, Y. Wang, P. J. Attayek, J. Balowski, X. F. Liu, R. J. Laurenza, L. T. Gaynor, C. E. Sims, J. A. Galanko, L. Li, N. L. Allbritton and S. T. Magness, Nat. Cell Biol., 2015, 17, 340–349 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14923a |
| This journal is © The Royal Society of Chemistry 2015 |