
Synthesis, characterization and photophysical properties of an acenaphthalene fused-ring-expanded NIR absorbing aza-BODIPY dye†
Abstract
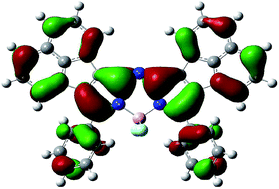
The synthesis and characterization of an NIR absorbing acenaphthalene fused-ring-expanded aza-BODIPY dye are reported. In contrast with its naphtho-fused analogue, a stable complex is obtained when the corresponding phthalonitrile is used as the precursor. A comparison is made with the photophysical properties of 3,5-diphenyl-aza-dibenzoBODIPY.
 Please wait while we load your content...
Please wait while we load your content...

