
Exploiting XPS for the identification of sulfides and polysulfides†
Abstract
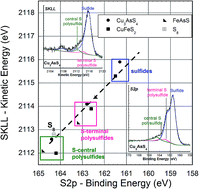
The identification of surface sulfide and polysulfide species based on the curve fitting of S2p photoelectron spectra and, for the first time, of X-ray excited S KLL Auger spectra has been performed. The different sulfur chemical states present on the surface (sulfide S2−, central S and terminal S in polysulfide chains) could be unambiguously assigned in the chemical state plot. Sulfur atoms in the central or terminal position, respectively, are found on a line with slope ca. −3 irrespective of the cation indicating similar initial state effects. On the other hand, for a given polysulfide, e.g. K2Sn, sulfur atoms both in central or terminal positions are found on the same line with slope −1 indicating similar final state effects. This behavior can be rationalized with the fact that the negative charge in polysulfide chains is located mainly on sulfur atoms in the terminal position; indeed, sulfur present as central S shows a binding energy shift of −0.6 eV with respect to elemental sulfur (S8), and sulfur in terminal S a shift of −2.4 eV. An application of this approach tested on commercial alkali polysulfides is provided for the curve fitting of SKLL signals and sulfur speciation of three different sulfide minerals enargite (Cu3AsS4), chalcopyrite (CuFeS2) and arsenopyrite (FeAsS). Also for the surface of mineral sulfides, terminal S atoms and central S atoms in the polysulfide chains can successfully be identified.
 Please wait while we load your content...
Please wait while we load your content...

