
Tuning SEI formation on nanoporous carbon–titania composite sodium ion batteries anodes and performance with subtle processing changes†
Jeongwoo Leea,
Yu-Ming Chenb,
Yu Zhub and
Bryan D. Vogt*a
aDepartment of Polymer Engineering, University of Akron, Akron, OH 44325, USA. E-mail: vogt@uakron.edu
bDepartment of Polymer Science, University of Akron, Akron, OH 44325, USA
First published on 10th November 2015
Abstract
The morphology of composite materials used in battery electrodes is critical to provide the requisite transport paths for ions and electrons to enable high performance. In this work, we describe a simple and scalable method to fine tune the morphology of carbon/TiO2 composite through polymerization-induced phase separation of a mixture containing commercial TiO2 nanoparticles, poly(hydroxyethyl methacrylate) (PHEMA), and photoacid generator (PAG) dissolved in furfuryl alcohol (FA, monomer). UV exposure converts the PAG to a strong acid that catalyzes the FA polymerization to quickly initiate the polymerization. The morphology is modulated by the molecular weight of PHEMA and FA concentration that impact the miscibility and mobility during phase separation. The polymerized composite is carbonized to yield porous carbon/TiO2 electrodes. The cycling performance is dictated by the morphology that develops during phase separation. Electrochemical impedance spectroscopy (EIS) analysis illustrates that subtle changes in synthetic conditions can dramatically impact the electrical or ion conductance, primarily through modulation in the solid electrolyte interphase (SEI). A careful investigation of the SEI layer on the porous carbon/TiO2 composites demonstrates a clear correlation between the SEI and the surface area of the porous anode as determined by transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS). With selection of synthetic conditions to yield a modest surface area composite, sustainable anodes with stable capacity can be fabricated for use in Na ion batteries.
Introduction
Lithium-based batteries have dominated non-stationary energy storage devices on the basis of their high energy density and capacity associated with their low weight, low redox potential, and ease of insertion.1–4 However, one of the critical challenges for lithium-based batteries is cost,5 which is partially driven by the high extraction costs of lithium metal and its limited natural abundance.6,7 Unlike lithium, sodium is one of the most naturally abundant elements (6th most common) with a crustal concentration estimated to be 3 orders of magnitude greater than lithium.7 This abundance has recently led a resurgence in sodium-ion battery investigations, despite two major drawbacks compared to lithium. First, the increased size of sodium (1.06 Å) compared to lithium ion (0.76 Å) limits the intercalation rate of Na+ and associated diffusion processes.8 Second, the molar mass of sodium (23 g mol−1) is 3 times more than that of lithium (6.9 g mol−1), but both ions carry the same charge (+1); this produces a significant decrease in the specific capacity on a mass basis for sodium.7 However in applications where cost dominates weight restrictions, such as grid energy storage, and in emergent markets, sodium ion batteries provide significant promise for energy storage.Due to its low cost and abundance, hard carbon is a commonly used anode materials for sodium ion batteries.9–11 A rate capacity between 100 and 300 mA h g−1 has been generally obtained for hard carbons, with the capacity dependent on both the allotrope and morphology,7,12–14 although Ponrouch et al.11 have recently demonstrated a hard carbon anode with capacity surpassing 320 mA h g−1. One intrinsic limitation associated with carbon is its limited charge storage on a volumetric basis (low mass density). Therefore, alternative materials for sodium ion battery anodes have been examined to obtain higher capacity. In particular, titanium dioxide (TiO2) has been studied as an anode material in sodium ion batteries8,15–19 due to its low cost, long cycle life, and ecofriendliness.8,15–19 However, the electrical conductivity of TiO2 is low. Therefore, composite anode materials for sodium ion batteries that combine the merits of carbon and TiO2 have been extensively investigated.20,21 For example, Yang et al.22 fabricated N-doped TiO2 nanorods decorated with carbon dots with capacity of ∼250 mA h g−1, and carbon-coated TiO2 nanoparticles by Jiang et al.23 exhibited a capacity of ∼240 mA h g−1. Alternative composites based on nanosized binary elemental alloys such as SnSb/C (435 mA h g−1)24 and Sn–SnS–C (664 mA h g−1)25 can provide further improvements in the capacity, but at the expensive of cost and environmental impact.
A significant challenge with the development of high performance composite electrodes for sodium ion batteries is to understand how the morphology impacts the performance.26 Ionic and electronic conduction pathways are critical to enable high performance, while high surface area electrodes imply a high contact area with the electrolyte, which should ease the ions flux across the electrode/electrolyte interface.27,28 However, the long term performance of high surface area electrodes is limited in many cases by the generation of the solid electrolyte interphase (SEI) layer. The SEI layer formation consumes electrolyte and alkali metal ions29 and tends to result in an irreversible capacity loss during the initial charge cycle. However, the SEI layer is generally electronically insulating and ionically conducting to protect the electrolyte from further decomposition if the SEI is stable.30 Agubra et al. have demonstrated that the electrolyte decomposition and subsequent SEI layer formation on an anode electrode increases the impedance in lithium ions.31 Recently, analysis of the SEI formation on Na2Ti3O7 anodes revealed the reaction between a standard binder (polyvinylidene difluoride) and the electrolyte that leads to an unstable SEI layer.32 However, the interplay between morphology and SEI layer formation in composite electrodes has not been thoroughly investigated for sodium ion batteries.
Here, we investigate this interplay for carbon/TiO2 composite anode electrodes for sodium ion batteries. The reversible capacity of these anodes for sodium ion batteries (<130 mA h g−1) is inferior to the state-of-the-art for carbon/TiO2 (e.g. 155 mA h g−1 for microsphere C–TiO2 (ref. 33) and 242 mA h g−1 for carbon-coated TiO2 nanoparticles23), but the fabrication of these composites is simple and tunable, based on polymerization-induced phase separation using inexpensive and commercially available materials. This method enables identification of key morphological aspects that impact the performance of the composites. In this case, commercial TiO2 nanoparticles are dispersed in furfuryl alcohol solution containing poly(hydroxyethyl methacrylate), PHEMA, and a photoacid generator (PAG). UV exposure generates a superacid to induce polymerization to poly(furfuryl alcohol), PFA; PFA provides the carbon matrix of non-graphitizing char,34 while PHEMA fully decomposes to yield pores during carbonization. The morphology of these porous carbon/TiO2 composites can be readily controlled using the molecular weight of the PHEMA and relative concentration of components. This simple route to modulate the morphology enables improved understanding of the SEI formation in these composites for sodium ion batteries.
Experimental
Materials
Furfuryl alcohol (FA, 98%), ethyl alcohol (EtOH, ≥99.5%), N-methyl-2-pyrrolidone (NMP, 99.5%), and poly(vinylidene fluoride) (PVDF, Mn = 107 kDa) were obtained from Sigma-Aldrich and used as received without further purification. Poly(2-hydroxyethyl methacrylate) (PHEMA) with nominal Mw = 20 kg mol−1 (20k), 300 kg mol−1 (300k), and 1000 kg mol−1 (1000k) was purchased from Scientific Polymer Products, Inc. (Ontario, NY) and used as received. Titanium oxide (TiO2, anatase, 15 nm) was purchased from Nanostructured & Amorphous Materials, Inc. The TiO2 nanoparticles were separated from water by centrifugation (accuSpin™400, Fisher Scientific) at 7000 rpm for 5 min, and the highly concentrated TiO2 slurry was re-dispersed into deionized water and EtOH using an ultrasonic cleaner (VWR® symphony™, operating frequency: 35 kHz, VWR International) to remove the surfactant dispersant and then collected again by centrifugation. This washing process was repeated 3 times. Rhodorsil PI2074 (CAS#: 178233-72-2) was obtained from Promerus, LLC (Brecksville, OH) and used as the photoacid generator. Sodium perchlorate (NaClO4, 98%, Sigma Aldrich) dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of ethylene carbonate (EC, 99%, Sigma Aldrich) and propylene carbonate (PC, 99.7%, Sigma Aldrich) was used as an electrolyte for battery testing. Na metal (99.8%, Acros Organics) was used as the counter electrode and copper foil (0.025 mm thick, Puratronic®) was used as a current collector for the anode electrode in the coin cells.
:1 (v/v) mixture of ethylene carbonate (EC, 99%, Sigma Aldrich) and propylene carbonate (PC, 99.7%, Sigma Aldrich) was used as an electrolyte for battery testing. Na metal (99.8%, Acros Organics) was used as the counter electrode and copper foil (0.025 mm thick, Puratronic®) was used as a current collector for the anode electrode in the coin cells.
Fabrication of PHEMA/PFA/TiO2 composites
In order to fabricate the composites, 0.35 g of PHEMA was dissolved in a mixture of FA (0.2, 0.3, 0.5, or 0.7 g) and 0.2 g EtOH. 0.3 g of TiO2 was ultrasonically dispersed in 0.3 g EtOH and subsequently 1.5 mg of Rhodorsil PI2074 was dissolved in this suspension. This mixture was combined with the polymer solution and cast into a PTFE Petri dish to a thickness of ∼0.2 mm. FA polymerization was induced by broadband UV (Spectroline, 4500 μW cm−1) exposure and polymerization was carried out at 60 °C for 10 min. The composite was pyrolyzed at 900 °C in N2. The heating schedule was initially 1 °C min−1 to 650 °C with 3 h hold at 650 °C, 1 °C min−1 to 900 °C, and then hold at 900 °C for 1 h. The temperature was then decreased at 3 °C min−1 to room temperature.Characterization
The morphology was characterized by transmission electron microscopy (TEM, JSM-1230, 120 kV, JEOL). Cross-sections of the composites, approximately 70 nm thick, were prepared by ultramicrotome (PT-PC PowerTome, RMC) using a diamond knife. To enable microtomed samples, the composite was embedded in epoxy resin (Embed-812 Resin, Electron Microscopy Sciences) that was hardened at 60 °C for 12 h. The specimens were supported on a 3.05 mm diameter Cu grid (01753-F, TED PELLA, Inc.). The mobility of precursor during FA photopolymerization is estimated by the suspension viscosity (Rotonetic™ 2 drive, Bohlin Gemini) through the initial stages of polymerization. During the measurement of viscosity, OmniCure® S2000 (200 W Mercury Arc, Lumen Dynamics) is used as the UV light source for in situ FA photopolymerization. The pore texture was elucidated by nitrogen adsorption–desorption isotherms (Micromeritics TriStar II instrument) at 77 K to determine the specific surface area by the Brunauer–Emmett–Teller (BET) method. The composition of the composite was estimated from thermogravimetric analysis, TGA Q50 (TA Instrument), with a heating rate of 20 °C min−1 to 700 °C in air. To avoid excess water in the sample, the composite materials were dried overnight under vacuum at 85 °C. All mass loss was attributed to removal of carbon. Powder X-ray diffraction (Ultima IV, Rigaku, Japan) was used to confirm the crystal structure of TiO2 particles. The diffractometer used Cu Kα radiation at 40 kV and 35 mA. The diffraction data was measured from 20° < 2θ < 60° with a step size of 0.04° at 1° min−1. The XRD profiles were analysed for anatase and rutile content using the relative intensity ratio (RIR) method:where Iα and Iβ are characteristic peak intensities of α and β phases, Wα and Wβ are weight fractions of α and β respectively. K is a constant determined by calibration curve with known samples.35 In this case, the content was calculated using PDXL2 software (Rigaku Corporation, v2.1.3.4) with the RIR values for the anatase and rutile of 4.99 and 3.62. The chemical composition of the neat and charge–discharge cycled carbon/TiO2 composite anodes was probed with X-ray photoelectron spectroscopy (XPS, PHI 5000 Versa probe II scanning XPS microprobe, ULVAC-PHI, Inc.) at a takeoff angle of 45 using pass energy of 11.75 eV at 2 × 10−6 Pa. High resolution scans at energies associated with C1s and O1s were performed to understand differences in the oxidation states. Each spectrum was collected using a monochromatic (Al Kα) X-ray beam (1486 eV) over a 200 μm diameter probing area. The peaks in the XPS spectra were fit using PHI MultiPak software.
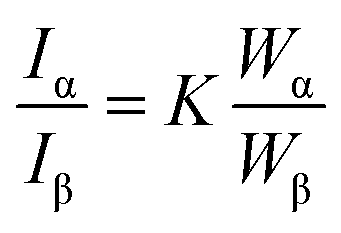
Battery fabrication and testing
The carbonized composite materials were ground by mortar and pestle into a powder and mixed with carbon black (N330, Sid Richardson Co.), and PVDF (binder) at 85:5:10 (w/w/w) ratio for solids. A small amount of NMP (∼30–40 mg to ∼0.13 g of the carbonized composite powder) was added to generate concentrated slurry. The slurry was coated onto 13 mm (diameter) punched copper foil using a Mayer rod (RDS 22) and the coated foil was dried overnight in vacuum at 80 °C. Representative anodes contained approximately 1.5 mg of active material. CR2032 coin cells (MTI Corporation) were assembled in an Ar-filled glove box (O2 < 0.5 ppm, H2O < 0.5 ppm) using the composite on copper foil as the anode electrode, Na metal as the cathode electrode, Celgard® 3501 (Celgard, LLC.) as the separator, and 1 M solution of NaClO4 in EC and PC (1:1 v/v) as the electrolyte.
The performance of the coin cells was determined using galvanostatic charge and discharge experiments with a battery tester (BST8-WA, MTI) at current densities between 10 and 50 mA g−1. The potential window used was 0.01–2.00 V vs. Na/Na+, consistent with prior studies for Na-ion batteries using TiO2 as the active anode.7,16,21 Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) analysis were carried out in CR2032 coin cells using an electrochemical workstation CHI660D (CH Instruments). The CV was performed using a potential range of 0.01–2.0 V at a scan rate of 0.1 mV s−1, and the EIS was conducted with applied amplitude of 5 mV in the range of 100 kHz to 0.01 Hz, generally after cycling the electrodes for 30 or 40 charge–discharge cycles.
For the SEI layer characterization, the cycled coin cells were carefully opened using a disassembling machine (MSK-110D, MTI), and the carbon/TiO2 composite electrodes were briefly rinsed by propylene carbonate and subsequently ethanol. The rinsed electrodes were dried in a vacuum oven at 80 °C before analyses. A reference as-produced electrode was prepared for comparison to the cycled electrodes. These reference electrodes were produced using the same concentrated slurry with carbon/TiO2 composite, carbon black, PVDF, and NMP. Additionally, these control electrodes were assembled as coin cells with sodium metal, separator, and electrolyte, but no potential was applied to the control electrodes. The same disassembly and cleaning procedure was used for the control and cycled electrodes.
Results and discussion
Fig. 1A illustrates the N2 adsorption–desorption isotherms for the carbon/TiO2 composites fabricated with varying molecular weight of PHEMA. These materials all exhibit type IV isotherms with a hysteresis loop at high relative pressures, P/P0.36 For the small molecular weight PHEMA, the hysteresis loop closes near P/P0 ∼ 0.9, which suggests the primary pores are connected by large mesopore windows. Conversely, the composite fabricated with the high molecular weight (1000k) PHEMA exhibits a much broader hysteresis loop that does not close until P/P0 ∼ 0.4. This suggests a broad distribution in window size between primary pores with small mesopores connecting the larger pores. The surface area of these composites increases with the molecular weight of PHEMA from 43.0 (20k) to 63.8 m2 g−1 (1000k), which corresponds with changes in morphology of the carbon/TiO2 composites (Fig. S1†). The pores of the carbon/TiO2 composite are significantly larger for the composite produced using 20k PHEMA (Fig. S1A†) than the carbon/TiO2 composites from 300k (Fig. S1B†) and 1000k PHEMA (Fig. S1C†). The morphology of porous carbon/TiO2 composites pyrolyzed from PHEMA/FA/TiO2 appears to be controlled by the mobility of the precursor suspension that decreases as the molecular weight of PHEMA increases. To confirm the lower mobility in the high molecular weight precursor suspensions, the viscosity of the solution during FA polymerization is used as a surrogate for mobility (Fig. S2†). Although the viscosity of all solutions increases during polymerization, a much larger viscosity and increase in viscosity as polymerization proceeds are found for the higher molecular weight of PHEMA. Additionally, the TiO2 nanoparticles appear to be more aggregated in the composites formed with higher molecular weight of PHEMA. For the composites fabricated with higher molecular weight PHEMA, the distribution of TiO2 nanoparticles appears to become increasingly more heterogeneous with the nanoparticles preferentially segregated near the pore interface.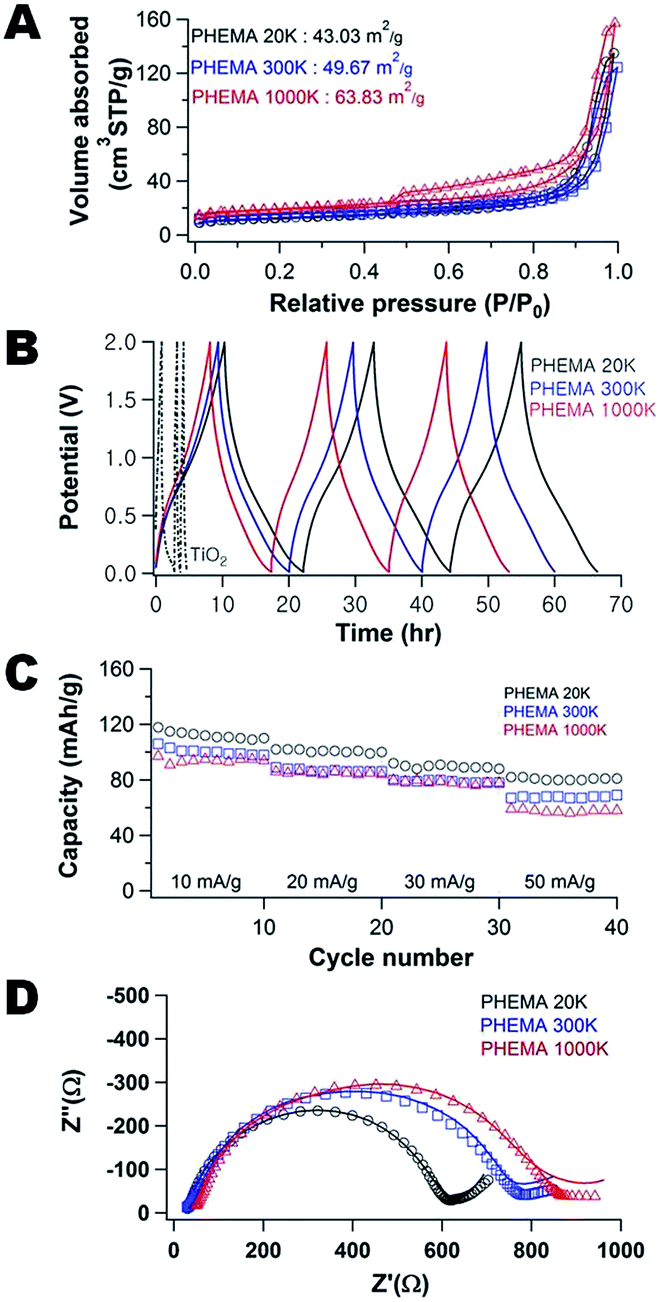 | ||
| Fig. 1 (A) N2 adsorption and desorption isotherms, (B) galvanostatic charge–discharge cycling for the composite electrodes at 10 mA g−1, (C) impact of rate on capacity during galvanostatic cycling, and (D) Nyquist plots of the porous carbon/TiO2 composite fabricated using 0.35 g PHEMA/0.3 g FA/0.3 g TiO2 as a function of molecular weight of PHEMA: 20k (○), 300k (□), and 1000k (Δ). | ||
Fig. 1B illustrates the galvanostatic charge–discharge curves for pure TiO2 (includes carbon black) and the carbon/TiO2 composite. First, the capacity of the TiO2 electrode is only approximately 20 mA h g−1 on the first discharge and this fades to less than 10 mA h g−1 after only 3 cycles. This poor cycling performance of the TiO2 anode is attributed to structural pulverization during the insertion/desertion of sodium.37 The composite fabricated with 20k PHEMA exhibits a much larger initial capacity of approximately 120 mA h g−1 and the fade is almost negligible in comparison to the pure TiO2 electrode. Additional galvanostatic charge discharge curves are shown in Fig. S3 and S4.† To better illustrate the cycling stability of the composite electrodes, Fig. 1C shows the impact of molecular weight of PHEMA used in the fabrication of the anode on the capacity of the porous carbon/TiO2 composite anodes for a range of charge–discharge rates. Although high surface area generally improves the capacity by increasing the flux across the electrode/electrolyte interface,27 the capacity of these porous carbon/TiO2 composite anodes decreases with increasing surface area. This behavior is somewhat surprising as the capacitive fade on cycling is quite small as shown in Fig. 1B. The capacity is stable for all materials examined after approximately 10 cycles. Increasing the current density from 10 mA g−1 (about 0.1C) up to 50 mA g−1 (about 0.8C) decreases the capacity for all samples; however, the highest capacity is always found for the composite fabricated with the lowest molecular weight PHEMA. However, the capacity of these electrodes is less than one might expect from anatase TiO2.8,15–19 The high temperature associated with carbonization of the PFA could also lead to conversion of the anatase42 to rutile43 phase. As determined from XRD (Fig. S5†), the rutile fraction in the TiO2 increases from 1.7% to 10.4% after carbonization of PFA/PHEMA/TiO2 composite The capacity of rutile has been demonstrated to be inferior to anatase for sodium ion batteries,8 so the lower capacity performance is not unexpected.
Fig. 1D shows the EIS spectra of the carbon/TiO2 composite electrodes after 40 charge–discharge cycles as a function of the molecular weight of PHEMA used in their fabrication. These spectra provide insight into the surface (ion adsorption and desorption) and bulk (ion diffusion) processes associated with charge storage.38 Nyquist plots for these electrodes exhibit a semi-circle at high frequency and then a linear increase at low frequency associated with the Na+ ion diffusion in the anode material (Warburg impedance).39 As the molecular weight of PHEMA increases, the diameter of semi-circle increases (increased the charge transfer resistance9). Additionally, the ohmic resistance, R, associated with the bulk carbon, grain boundary and interface resistances determined at high frequency (Z′ ∼ tens of Ω) increases slightly as the molecular weight of the PHEMA used in the fabrication increases. This behavior is somewhat unexpected as the composition for the electrodes are identical. This increased RΩ suggests that the extent of the SEI formation is dependent on the pore morphology. The higher surface area electrodes exhibit increased resistance from both R and the SEI layer. At low frequency after the semi-circle, the impedance is associated with the semi-infinite diffusion of the Na+ ions. However, the impedance for the composite fabricated with the highest molecular weight PHEMA behaves unexpectedly with Z′′ nearly independent of Z′. This decrease of Warburg slope (semi-infinite diffusion) as a function of the molecular weight of PHEMA is attributed directly to the diffusion limitations of the system.40 To better quantify the differences in the structure of the composite electrodes, the EIS data were fit to an electric circuit model (Fig. S6†). The bulk resistance (includes electrolyte, separator and electrode) increases from 3.9 W to 15.0 W to 24.5 W as the molecular weight of PHEMA used in the fabrication increases. Additionally, the resistance attributed to the formation of the SEI layer is increased by nearly an order of magnitude for the electrode fabricated with 1000k PHEMA in comparison to using the 20k PHEMA. The SEI layer appears to evolve through the first 10 charge–discharge cycles (Fig. S7). The formation of the SEI layer is further investigated through cyclic voltammetry as shown in Fig. S8.† Redox peaks were observed between 0.01 and 1.0 V. These peaks are attributed to the insertion and extraction of sodium ion at the carbon/TiO2 composite.41 In the first cathodic scan, there are current peaks at potentials greater than 1.0 V, whereas these peaks fade in the second and subsequent scans. This evolution is attributed to the irreversible formation of the SEI layer.41 The charge/discharge cycle appears to be stabilized after 10 cycles.
To better understand the origins of the differences in impedance, Fig. 2 illustrates the morphology of the neat composite anodes (left) and after 40 charge–discharge cycles (right) as a function of molecular weight of PHEMA used in the fabrication. Consistent with the N2 sorption isotherms, the neat composite anodes contain smaller pores as the molecular weight of PHEMA used in their fabrication increases (Fig. S9†). We attribute this pore size dependence to the mobility during the FA polymerization (Fig. S2†). These differences impact how the morphology changes from the SEI layer as shown in Fig. 2. Smaller pore sizes lead to greater changes in the morphology. This behavior is consistent with SEI formation for some lithium ion electrodes where the decomposition is proportional to the surface area of the carbon electrode.44 The increase in dark spots in the TEM micrographs is attributed to the oxygen-rich SEI products (Na2CO3, R–OCO2Na, and R–CH2ONa).45,46 As shown on right panels in Fig. 2, the density of these dark spots increases with increasing surface area. The increased SEI on electrodes fabricated with high molecular weight PHEMA likely leads to the high impedance of the electrodes (Fig. 1D).
 | ||
| Fig. 2 TEM images of carbon/TiO2 composite anodes as-produced (left) and after 40 charge–discharge cycles (right) fabricated with (A) 20k, (B) 300k, and (C) 1000k PHEMA using 0.35 g PHEMA/0.3 g FA/0.3 g TiO2 for the precursor. | ||
XPS spectra provide complementary information about the SEI layer after 40 charge–discharge cycles (as shown in Fig. S10†). We select 40 cycles as the SEI layer is stabilized by this cycle. Table 1 illustrates the composition changes of C, O and Na between as-produced and cycled electrodes due to the formation of SEI layer on the surface of electrodes. As a result, the molar ratio of O and Na to C increases; for instance, the molar ratio of C:O:Na for the composite anode fabricated with 20k PHEMA changes from 1:0.3:0 to 1:1.6:1.1 because of the decomposition of the electrolyte (a solution of NaClO4 in EC and PC) and the formation of the passivating SEI layer from the breakdown of the electrolyte on the surface of the electrodes.
| PHEMA | As-produced | Cycled | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C% | O% | Na% | Ti% | Cl% | F% | C% | O% | Na% | Ti% | Cl% | F% | |
| 20k | 64.3 ± 1.7 | 20.1 ± 7.2 | 0 | 4.6 ± 2.1 | 0 | 16.7 ± 0.5 | 27.1 ± 1.1 | 42.7 ± 0.9 | 29.3 ± 1.0 | 0.1 ± 0.04 | 0.7 ± 0.04 | 0.2 ± 0.1 |
| 300k | 70.8 ± 4.8 | 16.3 ± 4.8 | 0 | 2.1 ± 0.2 | 0 | 10.2 ± 8.9 | 28.2 ± 0.8 | 43.9 ± 1.6 | 26.5 ± 0.5 | 0.1 ± 0.03 | 0.9 ± 0.1 | 0.4 ± 0.1 |
| 1000k | 82.7 ± 1.3 | 13.6 ± 0.2 | 0 | 1.2 ± 1.2 | 0 | 2.3 ± 1.4 | 36.7 ± 4.1 | 40.5 ± 1.0 | 22.0 ± 2.0 | 0.1 ± 0.04 | 0.5 ± 0.4 | 0.3 ± 0.2 |
To better understand the composition of the SEI layers, high-resolution XPS is used to investigate the 1s binding of C as shown in Fig. 3. The strong C1s peak at ∼284.6 eV is assigned to C–C bonds in the electrode and the C–C bonds are expected to be present in the carbon component of the neat composite. The oxygen-rich components associated with the SEI layer formed by electrolyte (EC and PC) decomposition are associated with the C–O–C bonds at ∼286.2 eV and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds at ∼289 eV.47,48 Intriguingly, as a function of the molecular weight of PHEMA used (20k vs. 1000k) in their fabrication, the peak intensity and peak area of O–CO decrease from 521C s−1 (44% of C area) to 330C s−1 (26.2% of C area), whereas the peak intensity and peak area of C–O–C increase from 212C s−1 (17.8% of C area) up to 274C s−1 (24.0% of C area). The increases of C–O–C peak intensity and area are attributed to the electrolyte (EC and PC) decomposition, where the CO bonds in EC and PC are reduced to C–O.49 The enhanced C–O–C bonds when the electrode is fabricated with higher Mn PHEMA is consistent with the increased SEI formation in the TEM micrographs.
O bonds at ∼289 eV.47,48 Intriguingly, as a function of the molecular weight of PHEMA used (20k vs. 1000k) in their fabrication, the peak intensity and peak area of O–CO decrease from 521C s−1 (44% of C area) to 330C s−1 (26.2% of C area), whereas the peak intensity and peak area of C–O–C increase from 212C s−1 (17.8% of C area) up to 274C s−1 (24.0% of C area). The increases of C–O–C peak intensity and area are attributed to the electrolyte (EC and PC) decomposition, where the CO bonds in EC and PC are reduced to C–O.49 The enhanced C–O–C bonds when the electrode is fabricated with higher Mn PHEMA is consistent with the increased SEI formation in the TEM micrographs.
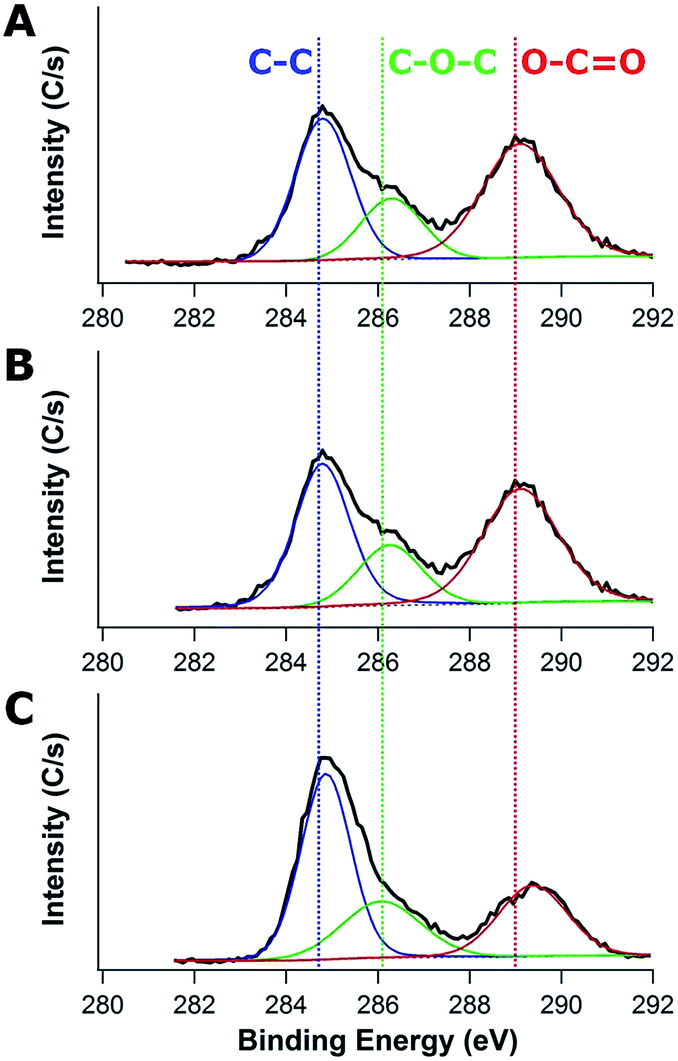 | ||
| Fig. 3 XPS spectra of carbon/TiO2 composite anodes after 40 charge–discharge cycles fabricated with (A) 20k, (B) 300k, and (C) 1000k PHEMA using 0.35 g PHEMA/0.3 g FA/0.3 g TiO2 for the precursor. | ||
The largest pore size leads to the best performance for the composites examined. The mobility of the PHEMA appears to determine the morphology of the composites (high mobility = larger pores) and thus 20k PHEMA is used for examining composition effects on performance. The carbon:TiO2 ratio can be easily modulated by the FA concentration in the precursor suspension. Fig. 4A illustrates how the N2 adsorption–desorption isotherms for the carbon/TiO2 composites are impacted by the concentration of FA used in their fabrication. At low concentration of FA (0.2 g), the surface area is at a maximum (74.1 m2 g−1), but then the surface area increases with increasing amount of FA in the precursor from a minimum of 43.0 (0.3 g) to 63.3 m2 g−1 (0.7 g). The increase in surface area can be rationalized from the changes in morphology of the carbon/TiO2 composites (Fig. S11†), where the pore size generally decreases as the FA concentration increases. The maximum in surface area at the lowest FA loading is attributed to exposed TiO2 nanoparticles in the composite (Fig. S11A†). These exposed nanoparticles provide roughness that increases the surface area.
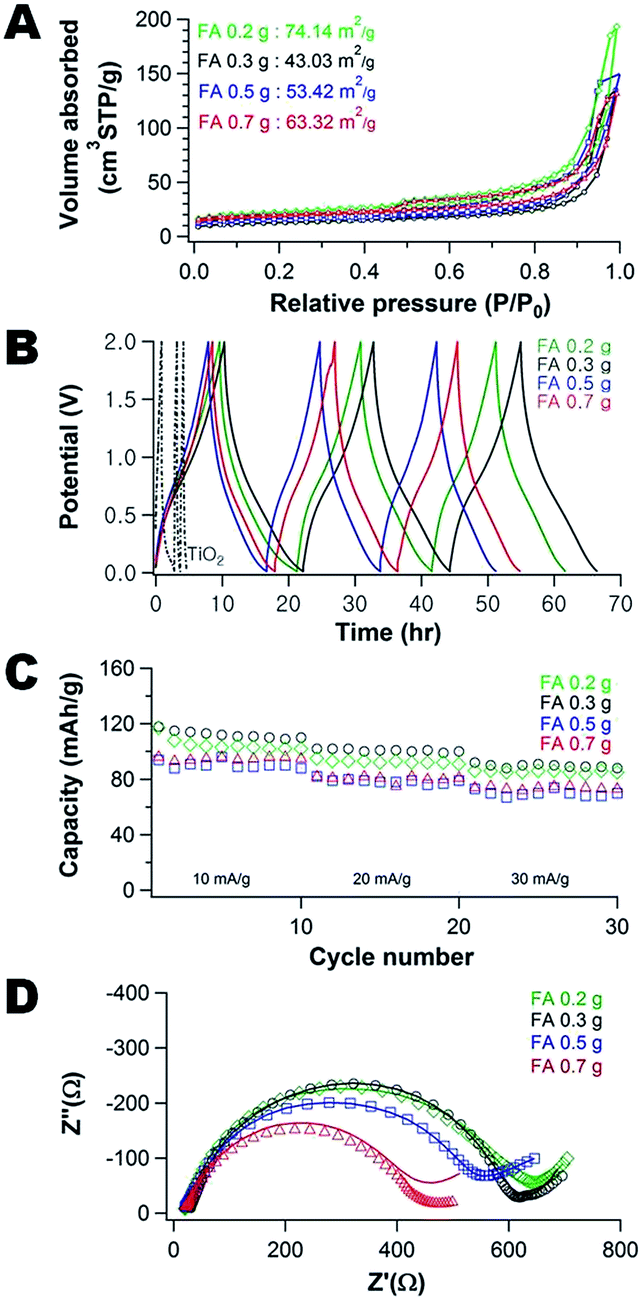 | ||
| Fig. 4 (A) N2 adsorption and desorption isotherms, (B) galvanostatic charge and discharge curves for the first 3 cycles, (C) galvanostatic cycling, and (D) Nyquist plots of the porous carbon/TiO2 composite fabricated using 0.35 g PHEMA (20k)/FA/0.3 g TiO2 as a function of amount of FA: 0.2 (◊), 0.3 (○), 0.5 (□), and 0.7 g (Δ). | ||
The pores of the carbon/TiO2 composite are significantly larger for the composite produced using 0.3 g FA (Fig. S11B†) than the carbon/TiO2 composites from 0.5 g FA (Fig. S11C†) and 0.7 g FA (Fig. S11D†). The Barrett–Joyner–Halenda (BJH) average pore sizes of the different composites using 0.3 g, 0.5 g, and 0.7 g of FA are ∼24 nm, ∼16 nm, and ∼14 nm, respectively. For this series of samples, the TiO2 nanoparticles are mostly covered by the carbon produced from the FA and thus the surface area scales inversely with pore size as expected (as the volume adsorbed is similar between the samples from Fig. 4A). Thus, the morphology of porous carbon/TiO2 composites pyrolyzed from PHEMA/FA/TiO2 can also be controlled by the amount of FA used in the synthesis, in addition to the PHEMA molecular weight.
Fig. 4B illustrates the galvanostatic charge–discharge curves of the porous carbon/TiO2 composites as well as the neat TiO2 anode (containing carbon black). A significantly higher and more stable capacity is obtained from the composites in caparison to the neat TiO2 anode. Fig. 4C illustrates how the composition and morphology of the active carbon/TiO2 impacts the capacity stability. As the current density increases from 10 mA g−1 (about 0.1C) up to 30 mA g−1 (about 0.4C), there is a decrease in the capacity by approximately 25%, irrespective of the composition of the composite. Increasing the FA content generally leads to a decrease in performance. The difference in carbon content is relatively small with the carbon content increasing from 12 to 18 wt% for the composites using 0.2 and 0.7 g FA (see Fig. S12†). There is also a concurrent increase in surface area (consistent with molecular weight effect where surface area is inversely related to performance). The lowest FA content composite (0.2 g FA) exhibits a slightly lower capacity than the composite fabricated with 0.3 g FA; this material has the lowest carbon content (12 wt%) and the highest surface area, so the performance is anomalous for this series of materials. Two probable reasons are the limited carbon content leads to poor electrical conductivity and/or the difference in surface chemistry where more TiO2 nanoparticles are exposed, which could lead to a SEI layer that limits Na+ transport.
To better understand these differences in performance, the EIS spectra of the carbon/TiO2 composite electrodes are examined (Fig. 4D). As the carbon content increases, the impedance decreases at high frequency and the slope of the linear regions decreases at low frequencies. The Na+ ion diffusion into the electrode is hindered at the higher carbon content. To understand this behavior, the morphology and chemical changes of the cycled composite anodes are investigated by TEM and XPS.
The change of morphology between neat and cycled composites is shown in Fig. 5. The neat composite anodes (left side in Fig. 5) contain smaller pores as the amount of FA used in their fabrication increases (Fig. S13†). The FA content controls the amount of carbon in the composites after pyrolysis of PHBMA/PFA/TiO2. From the TEM micrographs (right side in Fig. 5), it is possible to distinguish the low electron density carbon (light), the oxygen-rich SEI layers (darker regions) and the TiO2 nanoparticles (almost black). Thus generally, the SEI layer content increases as the carbon content of the composite materials increases. However, the lowest carbon content composite anode (Fig. 5A) appears to exhibit an increased SEI layer (dark spots) like composite anodes with higher carbon loading. We hypothesize that the exposed TiO2 nanoparticles lead to this increased SEI content.
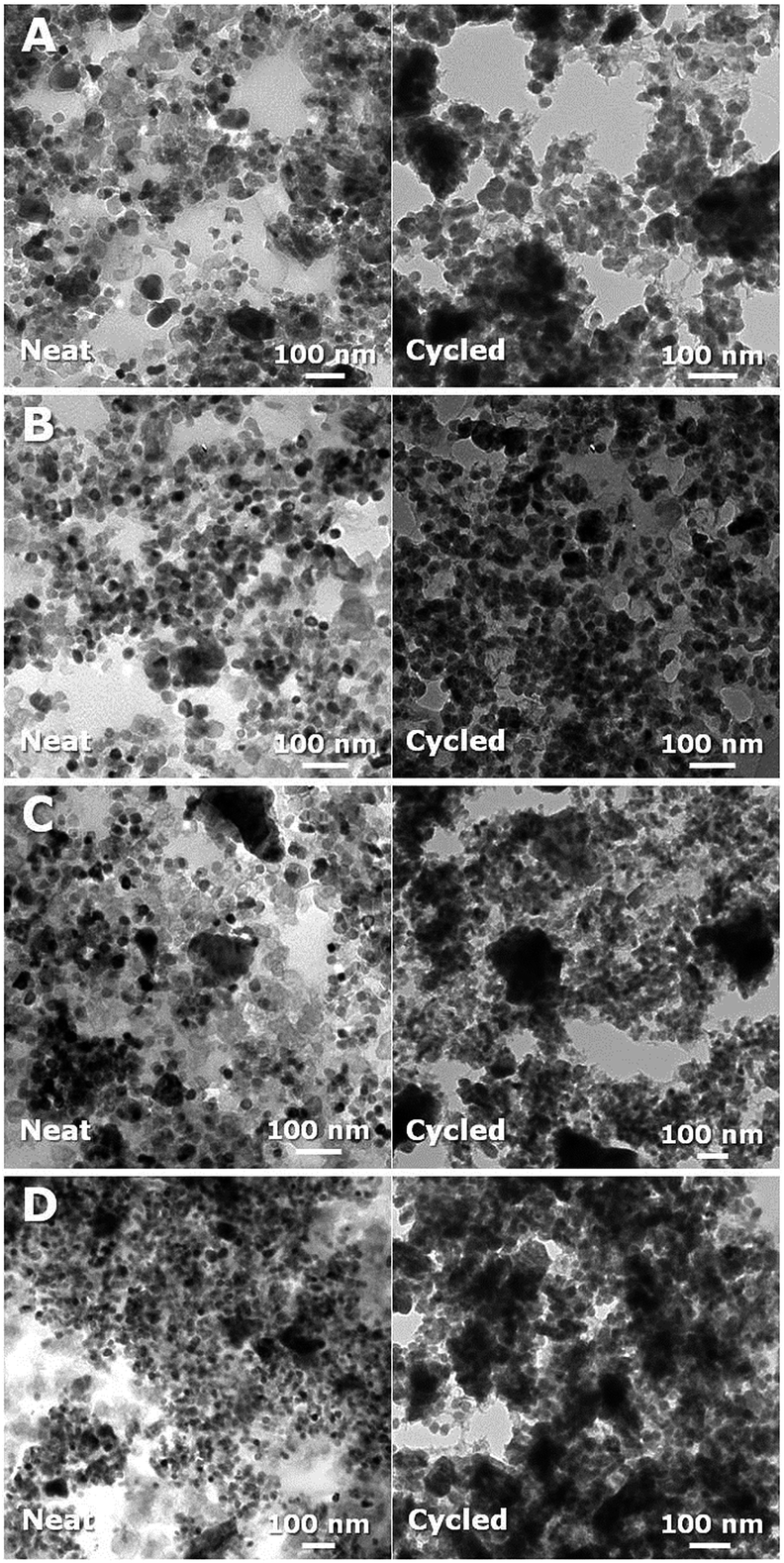 | ||
| Fig. 5 TEM images of carbon/TiO2 composite anodes as-produced (left) and after 30 charge–discharge cycles (right) fabricated with 0.35 g PHEMA (20k)/0.3 g TiO2/FA of (A) 0.2 g (12 wt% carbon), (B) 0.3 g (15 wt% carbon), (C) 0.5 g (16 wt% carbon), and (D) 0.7 g (18 wt% carbon). | ||
To further investigate the SEI layer formation, XPS spectra are obtained for the as-produced electrodes and after 30 charge–discharge cycles (Fig. S14†). Table 2 illustrates how the composition changes during cycling due to the formation of SEI layer. At low carbon content (12 wt%), the molar ratio of O and Na to C after 30 charge–discharge cycles increases much less than the other three anodes. We attribute this difference to the presence of exposed TiO2 nanoparticles in the low carbon content sample that leads to a different SEI layer, which is consistent with our hypotheses regarding the performance and impedance data for this sample.
| Carbon content | As-produced | Cycled | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C% | O% | Na% | Ti% | Cl% | F% | C% | O% | Na% | Ti% | Cl% | F% | |
| 12 wt% | 75.8 ± 0.9 | 17.8 ± 0.6 | 0 | 6.4 ± 0.3 | 0 | 0.2 ± 0.3 | 40.6 ± 10.2 | 35.6 ± 3.9 | 20.8 ± 6.2 | 0.1 ± 0.02 | 2.7 ± 0.6 | 0.2 ± 0.08 |
| 15 wt% | 64.3 ± 1.7 | 20.1 ± 7.2 | 0 | 4.6 ± 2.1 | 0 | 16.7 ± 0.5 | 27.1 ± 1.1 | 42.7 ± 0.9 | 29.3 ± 1.0 | 0.1 ± 0.04 | 0.7 ± 0.04 | 0.2 ± 0.1 |
| 16 wt% | 71.5 ± 7.8 | 20.6 ± 7.5 | 0 | 4.1 ± 3.1 | 0 | 6.8 ± 2.7 | 23.5 ± 2.7 | 41.4 ± 1.1 | 30.7 ± 1.9 | 0.2 ± 0.05 | 0.5 ± 0.4 | 0.6 ± 0.05 |
| 18 wt% | 75.2 ± 12.2 | 15.9 ± 11.2 | 0 | 4.8 ± 2.5 | 0 | 4.2 ± 3.1 | 37.7 ± 6.5 | 39.8 ± 2.7 | 21.2 ± 3.6 | 0.1 ± 0.01 | 1.2 ± 0.05 | 0.3 ± 0.1 |
High-resolution XPS spectra for the 1s binding of C and O are shown in Fig. 6. The area ratio of C–C bond to carbon–oxygen bonds (C–O–C and O–CO) decreases from 1:1.51 to 1:0.78 as the FA content increases, which corresponds to an increase in the carbon content of the anode from 15 wt% to 18 wt% (as shown in Table 3). This is unusual as the SEI layer appears to be more prevalent for the higher carbon content anodes from the TEM micrographs (Fig. 5), but there are less carbon–oxygen bonds near the surface relative to C–C bonds for the high carbon content anodes. This is counter to expectations based on the survey scan (Table 2). To better understand this behavior, high-resolution XPS spectra for O1s are investigated to identify CO (∼531 eV), C–O (∼532.5 eV), and C–OH (∼533 eV).50 As the carbon content increases (except for the low carbon, 12 wt%, outlier), the peak area of CO decrease from 64.6% to 52.1%, whereas the peak areas of C–O and C–OH increase from 10.9% to 27.1%, all on the basis of the total area for the oxygen peaks. These data are consistent reduction of the electrolyte (EC and PC) as the CO bonds are reduced to C–O.49
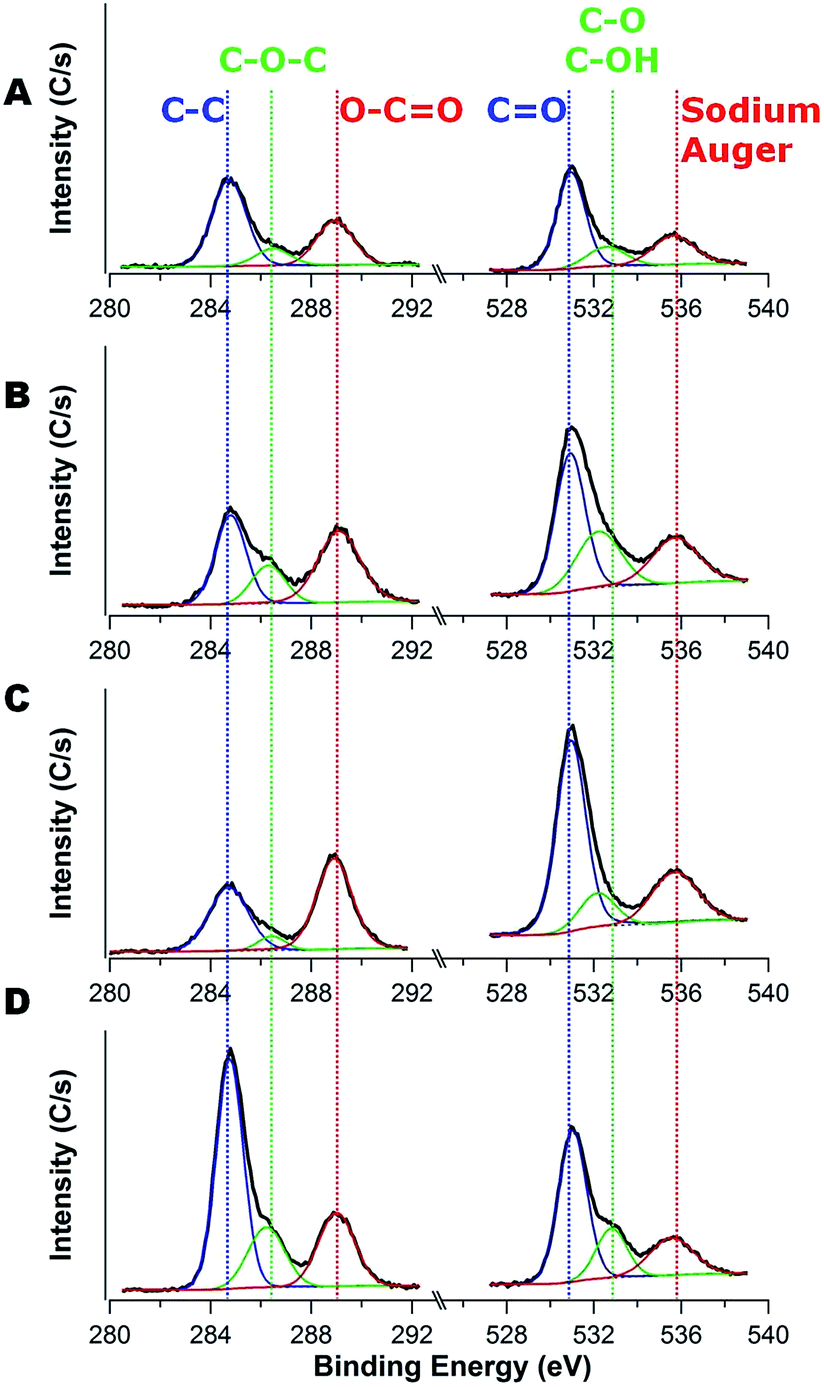 | ||
| Fig. 6 XPS spectra examining C1s (left) and O1s (right) for the anodes after 30 charge–discharge cycles fabricated with different carbon contents of carbon/TiO2 composite: (A) 12 wt%, (B) 15 wt%, (C) 16 wt%, and (D) 18 wt%. | ||
| Carbon content | C1s | |||
|---|---|---|---|---|
| C–C (∼284.6 eV) | C–O–C (∼286.2 eV) | O–CO (∼289 eV) |
C–C:(C–O–C + O–CO) |
|
| 12 wt% | 57.8 ± 0.6% | 11.1 ± 0.5% | 31.1 ± 0.1% | 1:0.73 |
| 15 wt% | 39.9 ± 2.2% | 21.4 ± 3.6% | 38.7 ± 1.5% | 1:1.51 |
| 16 wt% | 40.6 ± 0.5% | 6.3 ± 0.02% | 53.1 ± 0.5% | 1:1.46 |
| 18 wt% | 56.3 ± 0.01% | 19.5 ± 0.1% | 24.2 ± 0.01% | 1:0.78 |
| O1s | ||||
|---|---|---|---|---|
| CO (∼531 eV) |
C–O, C–OH (∼532.8 eV) | Na Auger (∼535.5 eV) | (CO + C–O, C–OH):Na Auger |
|
| 12 wt% | 58.8 ± 1.2% | 14.8 ± 1.6% | 26.5 ± 0.3% | 1:0.36 |
| 15 wt% | 64.6 ± 0.4% | 10.9 ± 1.0% | 24.8 ± 0.6% | 1:0.33 |
| 16 wt% | 63.9 ± 1.3% | 11.0 ± 2.1% | 24.6 ± 1.3% | 1:0.33 |
| 18 wt% | 52.1 ± 2.7% | 27.1 ± 3.2% | 20.9 ± 0.5% | 1:0.26 |
These XPS data focused on O1s provide an additional advantage to understand the SEI layer formation as the Na Auger is visible in the energy window examined. The nature of the SEI layer formed on carbons depends on the location: the basal plane, which consists of sp2 carbon atoms (C–C bonds), typically generates SEI that is rich in organic compounds, whereas the edge sites, which contain dandling bonds and various capping moieties (e.g., hydrogen, hydroxyl, carbonyl and carboxyl groups), typically generates SEI that is rich in inorganic compounds.51,52 Table 3 illustrates the change of the ratio between oxygen-rich substances (CO, C–O, and C–OH peaks) and sodium (Na Auger peak) as a function of the carbon content after 30 charge–discharge cycles. This ratio is the largest for the lowest carbon content (1:0.37) and decreases to a minimum at the highest carbon content (1:0.26). We attribute this difference to the propensity of organic-rich SEI to form on the basal plane of carbon, so the higher carbon content (18 wt%) should lead to additional SEI.
These results illustrate the interplay between surface area, composition, surface chemistry and pore size on determining the performance of anatase TiO2–carbon composite anodes for sodium ion batteries. In particular, these factors impact the SEI layer formation. Higher surface areas of the composite materials lead to additional SEI, while increasing the carbon content of the composite also generates more SEI. The SEI layer can adversely impact the long-term performance of the anodes with a decrease in capacity and an increase in impedance.
Conclusions
The morphology of porous carbon/TiO2 composites was tuned using the composition and molecular weight of the porogen in the precursor via a simple polymerization induced phase separation process using a renewable carbon source (furfuryl alcohol). The SEI layer formation increases on the anode having larger surface area, but the nature of the SEI appears to depend on minor perturbations in the composition of the anode. The composite with high carbon content has more sp2 carbon atoms (C–C bonds) and tends to produce a thick SEI layer that consists of primarily organic compounds (oxygen-rich substances). This high SEI layer formation limits sodium ion diffusion and impacts performance with an increase in the impedance and decreases in capacity as the carbon content in the composites increases. This simple polymerization induced phase separation method provides a facile route to teasing out how seemingly minor changes in composition and morphology impact the performance. This method could provide a route to help to better design electrode materials for sodium ion batteries by understanding interplay between structure and performance.Acknowledgements
This work has been partially supported by the National Science Foundation under grant CBET-1336057. Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund partial support of this research through award 53739-ND7. The authors acknowledge the assistance of Zhorro Nikolov with XPS and Bojie Wang with TEM measurements.References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- A. L. M. Reddy, S. R. Gowda, M. M. Shaijumon and P. M. Ajayan, Adv. Mater., 2012, 24, 5045–5064 CrossRef CAS PubMed.
- M. G. Kim and J. Cho, Adv. Funct. Mater., 2009, 19, 1497–1514 CrossRef CAS.
- A. Shcherbakova, A. Kleit and J. Cho, Appl. Energy, 2014, 125, 93–102 CrossRef.
- C. Bommier, W. Luo, W. Y. Gao, A. Greaney, S. Q. Ma and X. Ji, Carbon, 2014, 76, 165–174 CrossRef CAS.
- J. Lee, Y. M. Chen, Y. Zhu and B. D. Vogt, ACS Appl. Mater. Interfaces, 2014, 6, 21011–21018 CAS.
- D. Su, S. Dou and G. Wang, Chem. Mater., 2015, 27, 6022–6029 CrossRef CAS.
- T. Chen, Y. Liu, L. Pan, T. Lu, Y. Yao, Z. Sun, D. H. C. Chua and Q. Chen, J. Mater. Chem. A, 2014, 2, 4117–4121 CAS.
- W. Luo, J. Schardt, C. Bommier, B. Wang, J. Razink, J. Simonsen and X. L. Ji, J. Mater. Chem. A, 2013, 1, 10662–10666 CAS.
- A. Ponrouch, A. R. Goni and M. R. Palacin, Electrochem. Commun., 2013, 27, 85–88 CrossRef CAS.
- K. Tang, L. Fu, R. J. White, L. Yu, M.-M. Titirici, M. Antonietti and J. Maier, Adv. Energy Mater., 2012, 2, 873–877 CrossRef CAS.
- S. Wenzel, T. Hara, J. Janek and P. Adelhelm, Energy Environ. Sci., 2011, 4, 3342–3345 CAS.
- R. Alcantara, P. Lavela, G. F. Ortiz and J. L. Tirado, Electrochem. Solid-State Lett., 2005, 8, A222–A225 CrossRef CAS.
- Z. H. Bi, M. P. Paranthaman, P. A. Menchhofer, R. R. Dehoff, C. A. Bridges, M. F. Chi, B. K. Guo, X. G. Sun and S. Dai, J. Power Sources, 2013, 222, 461–466 CrossRef CAS.
- L. M. Wu, D. Buchholz, D. Bresser, L. G. Chagas and S. Passerini, J. Power Sources, 2014, 251, 379–385 CrossRef CAS.
- Z. Yan, L. Liu, J. Tan, Q. Zhou, Z. Huang, D. Xia, H. Shu, X. Yang and X. Wang, J. Power Sources, 2014, 269, 37–45 CrossRef CAS.
- L. Wu, D. Bresser, D. Buchholz, G. A. Giffin, C. R. Castro, A. Ochel and S. Passerini, Adv. Energy Mater., 2015, 5, 1401142 Search PubMed.
- H. Xiong, M. D. Slater, M. Balasubramanian, C. S. Johnson and T. Rajh, J. Phys. Chem. Lett., 2011, 2, 2560–2565 CrossRef CAS.
- D. Bresser, B. Oschmann, M. N. Tahir, F. Mueller, I. Lieberwirth, W. Tremel, R. Zentel and S. Passerini, J. Electrochem. Soc., 2015, 162, A3013–A3020 CrossRef CAS.
- H. A. Cha, H. M. Jeong and J. K. Kang, J. Mater. Chem. A, 2014, 2, 5182–5186 CAS.
- Y. Yang, X. Ji, M. Jing, H. Hou, Y. Zhu, L. Fang, X. Yang, Q. Chen and C. E. Banks, J. Mater. Chem. A, 2015, 3, 5648–5655 CAS.
- Y. Ge, H. Jiang, J. Zhu, Y. Lu, C. Chen, Y. Hu, Y. Qiu and X. Zhang, Electrochim. Acta, 2015, 157, 142–148 CrossRef CAS.
- L. Xiao, Y. Cao, J. Xiao, W. Wang, L. Kovarik, Z. Nie and J. Liu, Chem. Commun., 2012, 48, 3321–3323 RSC.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, J. Mater. Chem. A, 2013, 1, 7181–7184 CAS.
- C. Jo, Y. Park, J. Jeong, K. T. Lee and J. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 11748–11754 CAS.
- S. Goriparti, E. Miele, F. de Angelis, E. Di Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- Y. Ishii, Y. Kanamori, T. Kawashita, I. Mukhopadhyay and S. Kawasaki, J. Phys. Chem. Solids, 2010, 71, 511–514 CrossRef CAS.
- P. Arora, R. E. White and M. Doyle, J. Electrochem. Soc., 1998, 145, 3647–3667 CrossRef CAS.
- E. Markevich, V. Baranchugov, G. Salitra, D. Aurbach and M. A. Schmidt, J. Electrochem. Soc., 2008, 155, A132–A137 CrossRef CAS.
- V. Agubra and J. Fergus, Materials, 2013, 6, 1310–1325 CrossRef CAS.
- M. A. Munoz-Marquez, M. Zarrabeitia, E. Castillo-Martinez, A. Eguia-Barrio, T. Rojo and M. Casas-Cabanas, ACS Appl. Mater. Interfaces, 2015, 7, 7801–7808 CAS.
- S.-M. Oh, J.-Y. Hwang, C. S. Yoon, J. Lu, K. Amine, I. Belharouak and Y.-K. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 11295–11301 CAS.
- J. F. Mareche, D. Begin, G. Furdin, S. Puricelli, J. Pajak, A. Albiniak, M. Jasienko-Halat and T. Siemieniewska, Carbon, 2001, 39, 771–773 CrossRef CAS.
- H. Chen, S. Chen, L. Li and S. Jin, Propellants, Explos., Pyrotech., 2008, 33, 467–471 CrossRef CAS.
- K. Sing, Colloids Surf., A, 2001, 187, 3–9 CrossRef.
- K.-T. Kim, G. Ali, K. Y. Chung, C. S. Yoon, H. Yashiro, Y.-K. Sun, J. Lu, K. Amine and S.-T. Myung, Nano Lett., 2014, 14, 416–422 CrossRef CAS PubMed.
- S. B. Yoon, J. P. Jegal, K. C. Roh and K. B. Kim, J. Electrochem. Soc., 2014, 161, H207–H213 CrossRef CAS.
- S. Tanaka, A. Doi, T. Matsui and Y. Miyake, J. Power Sources, 2013, 228, 24–31 CrossRef CAS.
- R. P. Ramasamy, C. Feger, T. Strange and B. N. Popov, J. Appl. Electrochem., 2006, 36, 487–497 CrossRef CAS.
- D. Yan, C. Yu, Y. Bai, W. Zhang, T. Chen, B. Hu, Z. Sun and L. Pan, Chem. Commun., 2015, 51, 8261–8264 RSC.
- T. Chen, G. H. Guai, C. Gong, W. Hu, J. Zhu, H. Yang, Q. Yan and C. M. Li, Energy Environ. Sci., 2012, 5, 6294–6298 CAS.
- W. Hu, L. Li, W. Tong, G. Li and T. Yan, J. Mater. Chem., 2010, 20, 8659–8667 RSC.
- R. Fong, U. Vonsacken and J. R. Dahn, J. Electrochem. Soc., 1990, 137, 2009–2013 CrossRef CAS.
- L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J.-G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- J. T. Lee, N. Nitta, J. Benson, A. Magasinski, T. F. Fuller and G. Yushin, Carbon, 2013, 52, 388–397 CrossRef CAS.
- V. Eshkenazi, E. Peled, L. Burstein and D. Golodnitsky, Solid State Ionics, 2004, 170, 83–91 CrossRef CAS.
- Y. An, P. Zuo, C. Du, Y. Ma, X. Cheng, J. Lin and G. Yin, RSC Adv., 2012, 2, 4097–4102 RSC.
- P. Verma, P. Maire and P. Novak, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- W. Yuan, Y. Zhou, Y. Li, C. Li, H. Peng, J. Zhang, Z. Liu, L. Dai and G. Shi, Sci. Rep., 2013, 3, 2248 Search PubMed.
- K. Zaghib, G. Nadeau and K. Kinoshita, J. Power Sources, 2001, 97(8), 97–103 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM micrographs, representative galvanostatic charge–discharge and cyclic voltammetry curves, pore size distributions, and XPS data. See DOI: 10.1039/c5ra14907j |
| This journal is © The Royal Society of Chemistry 2015 |