
Synthesis of ketones via organolithium addition to acid chlorides using continuous flow chemistry†
Soo-Yeon Moon,
Seo-Hee Jung,
U. Bin Kim and
Won-Suk Kim*
Department of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea. E-mail: wonsukk@ewha.ac.kr; Fax: +82 2 3277 2384
First published on 14th September 2015
Abstract
An efficient method for the synthesis of ketones using organolithium and acid chlorides under continuous flow conditions has been developed. In contrast to standard batch chemistry, over-addition of the organolithium to the ketone for the formation of the undesired tertiary alcohol has been minimised representing a direct approach toward ketones.
Ketones are widely used structural motifs in the pharmaceutical and agrochemical industries as well as in natural product and synthetic chemistry.1 Traditionally, aryl ketones are prepared using Friedel–Crafts acylation,2 but this approach suffers from harsh reaction conditions, regioselectivity issues and the formation of side products.2c Another common method for the preparation of functionalised ketones involves the nucleophilic addition of organometallic reagents to carboxylic acid derivatives,3 but low yields are generally obtained without the use of additives such as ligands or Lewis acids due to over-addition to the ketone affording the undesired tertiary alcohol as the major product (Fig. 1A).4 In light of these short-comings, novel reagents derived from acid chlorides, such as N-methoxy-N-methylamides (Weinreb reagents),5a S-(2-pyridyl)thiolates,5b morpholine amides5c and tertiary amides,5d have been developed to enable acylation without over-addition (Fig. 1B). However, these methods require an additional acid chloride functionalisation step before yielding the ketone products, and while reactions such as organocuprate (Gilman reagent) addition to acid chlorides (Fig. 1C)6 and palladium-catalysed cross-coupling of carboxylic acid derivatives have been developed for the direct preparation of ketones under mild conditions (Fig. 1D),7 the need for expensive transition metal catalysts is also disadvantageous. Considering the continued interest in the preparation of ketones, it is surprising that the simple and straightforward addition of organolithium reagents to carboxylic acid derivatives have been scarcely reported in the literature.8
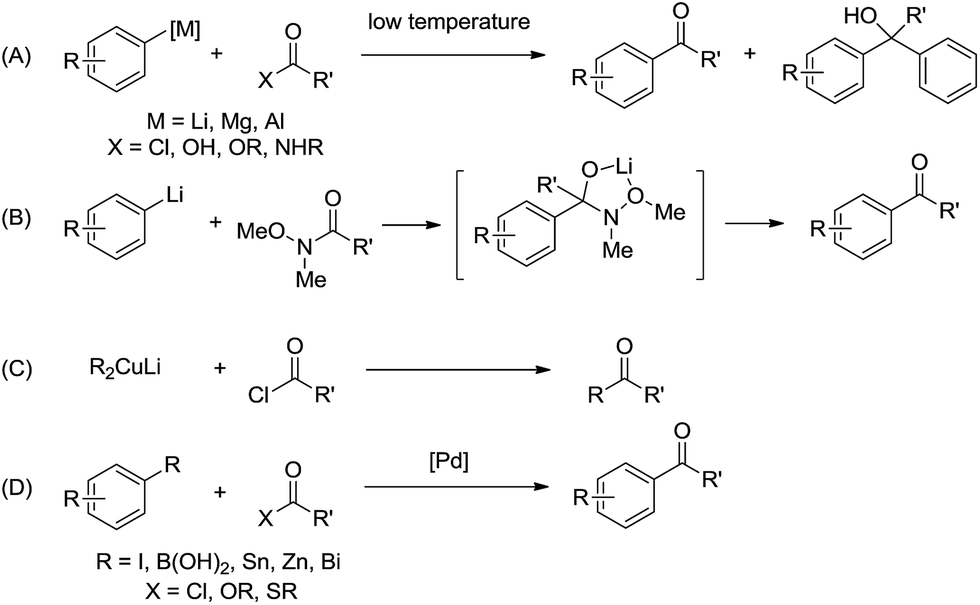 | ||
| Fig. 1 (A) Reaction of organometallic reagents with carboxylic acid derivatives. (B) Reaction of arene-lithiums with Weinreb reagents. (C) Reaction of Gilman reagent with acid chlorides. (D) Pd-catalysed cross coupling reaction with carboxylic acid derivatives. | ||
Important technological advancements in automation, mechanisation and micro-fluid reactor control have enabled a new approach to synthetic and medicinal chemistry termed continuous flow.9,10 Greater precision and control supported by machine-assisted chemistry enables the minimisation of waste, improved mass and heat transfers and controlled mixing. Due to these advantages inherent to flow reactors, chemistry not possible in batch settings can now be accessed using this “flash chemistry” coined by Yoshida et al.11 We hypothesised that the advantages afforded by the flow system could be exploited to react an organolithium reagent with an acid chloride to directly afford a flow-generated ketone, which would then be localised downstream and isolated away from further reaction with the organolithium reagents.
Recently, Jamison and co-workers described an efficient continuous flow synthesis of ketones from carbon dioxide and organolithium or Grignard reagents suppressing the undesired tertiary alcohol byproduct.12
With these considerations in mind, we now report a simple and efficient method for the direct synthesis of ketones without additives, using organolithium reagents and acid chlorides in a continuous flow reactor.
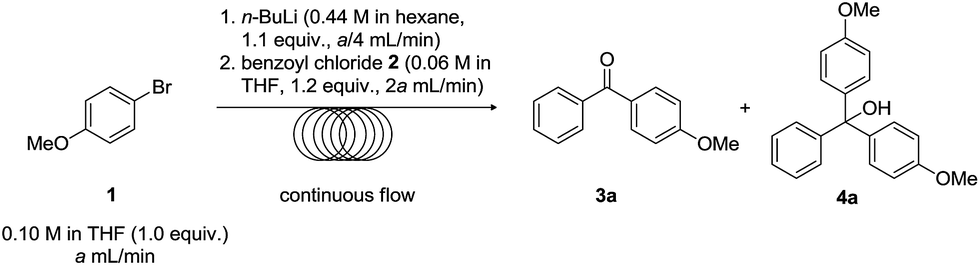
We chose to investigate the coupling between 1-bromo-4-methoxybenzene 1 and benzoyl chloride 2 as our initial trial. For comparison, the reaction was first run under standard batch conditions whereupon 1-bromo-4-methoxybenzene 1 was first lithiated with n-BuLi in THF at −78 °C for 1 h then quenched with benzoyl chloride 2 at −78 °C for 0.5 h. This afforded the desired ketone product 3a in trace yield, and as expected, afforded the undesired tertiary alcohol 4a in 72% yield with the aryl bromide being employed as the limiting reagent (Scheme 1A). To see if the results could be improved under inverse addition conditions, we repeated the reaction where we slowly added the lithiated 1-bromo-4-methoxybenzene 1 to solution of benzoyl chloride 2. However, no major differences were observed in terms of yield for this particular reaction. Similar results were obtained when using 2-methoxybenzoyl chloride 5 to quench lithiated 1-bromo-4-methoxybenzene 1 (Scheme 1B). When reaction with 1-bromo-4-methoxybenzene 1 was repeated under inverse addition conditions however, ketone 3d was isolated as the major product, in albeit more moderate yield (40%). Thus, although ketone could sometimes be isolated as the major product under batch conditions, the results are generally not satisfactory. For purposes of this study we did not optimise these reactions any further.
 | ||
| Scheme 1 Reaction between 1-bromo-4-methoxybenzene 1 and benzoyl chloride 2 under standard batch conditions. | ||
Having batch results in hand, we next moved to design our flow reactor. Our set-up was composed of three standard pressure syringe pumps and two miacromixers M1 and M2 (Fig. 2). A solution of 1-bromo-4-methoxybenzene 1 in THF (0.10 M) was pumped at a given flow rate (a mL min−1) into a micromixer M1 (Ø = 250 μm), where it was intercepted and mixed with n-BuLi in hexane (0.44 M) delivered by a second syringe pump at a rate quarter to the bromide solution (a/4 mL min−1).13 The newly formed organolithium stream was fed into micromixer M2, where it was mixed with a solution of benzoyl chloride 2 in THF (0.06 M) delivered by a third syringe pump at a rate double to the bromide solution (2a mL min−1) with a residence time RT. The newly formed ketone was subsequently collected and purified with the whole process having a collection time of 1.5 min at completion.
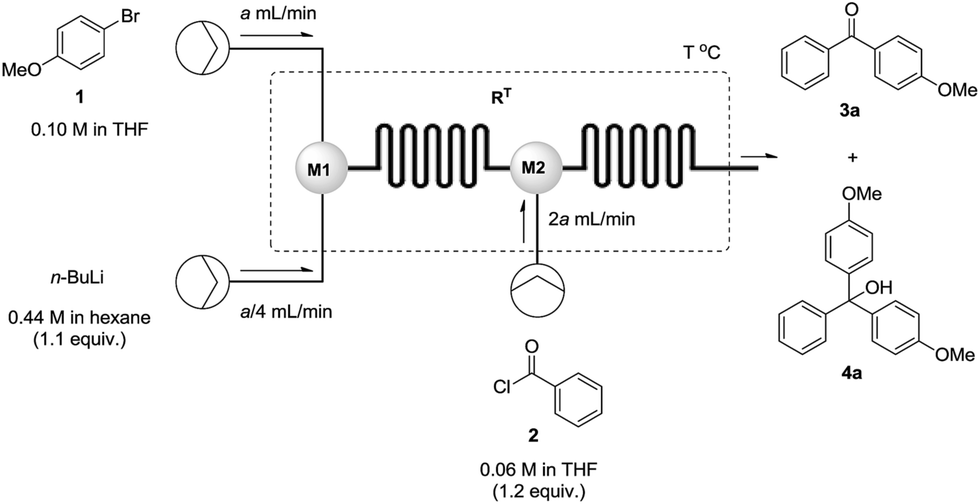 | ||
| Fig. 2 Schematic representation of the continuous flow set up used for optimisation of the reaction between 1-bromo-4-methoxybenzene and benzoyl chloride. | ||
As summarised in Table 1, the initial reaction of 1-bromo-4-methoxybenzene 1 with n-BuLi and benzoyl chloride 2 under flow conditions with an inner diameter of M1 (Ø = 250 μm) and M2 (Ø = 500 μm) and a flow rate a of 1 mL min−1 at 0 °C afforded only the undesired tertiary alcohol 4a in a low yield of 33% (entry 1). To prevent the undesired over-addition, we increased the flow rate a (entries 2–4) whereupon the desired ketone was formed in 21% yield at a flow rate a of 4 mL min−1 by virtue of the fast mixing and quick passage through the reactor (entry 4). However, the undesired tertiary alcohol was still a major side product (64%).
|
|||||
|---|---|---|---|---|---|
| Entrye | Flow rate a (mL min−1) | Inner diameter of M2 (μm) | Temperature (°C) | Yield of 3a c (%) | Yield of 4a c (%) |
| a Solvent for benzoyl chloride was CH2Cl2.b The concentration of acid chloride was increased to 0.075 M (1.5 equiv).c GC yield (internal standard was 4-chloroanisole).d Isolated yield.e VICI micromixer was used for M1 and M2 unless otherwise specified and the inner diameter of M1 was fixed as 250 μm.f YMC micromixer was used for M1 and M2.g ITEC micromixer was used for M2. | |||||
| 1 | 1 | 500 | 0 | 0 | 33 |
| 2 | 2 | 500 | 0 | 11 | 64 |
| 3 | 3 | 500 | 0 | 17 | 68 |
| 4 | 4 | 500 | 0 | 21 | 64 |
| 5f | 4 | 300 | 0 | 33 | 60 |
| 6 | 4 | 250 | 0 | 51 | 42 |
| 7 | 4 | 250 | −20 | 54 | 37 |
| 8 | 4 | 250 | −40 | 57 | 33 |
| 9 | 4 | 250 | −78 | 55 | 28 |
| 10a,b | 4 | 250 | −40 | 73(66d) | 19(13d) |
| 11b,f | 4 | 250 | −40 | 71 | 19 |
| 12b,g | 4 | 250 | −40 | 71 | 18 |
Progressive reduction of mixer M2's inner diameter (Ø) from 500 μm to 250 μm (entries 5 and 6) revealed M2 (Ø = 250 μm) to be the most optimal inner diameter for the best conversion affording 51% of ketone 3a and 42% of undesired tertiary alcohol 4a. To further limit the over-addition side product, the reaction temperature was controlled. As highlighted in entries 7–9, a reduced temperature of −40 °C afforded the desired ketone in better yield (entry 8, 57%), while there were no significant improvement when the temperature was reduced further (e.g., entry 9, −78 °C). Finally, we switched the delivery solvent for the benzoyl chloride from tetrahydrofuran to dichloromethane and increased its concentration to 0.075 M at −40 °C to propitiously afford the desired ketone in 66% isolated yield and 13% of undesired tertiary alcohol (entry 10). Use of different micromixers such as YMC (entry 11) and ITEC (entry 12) afforded the desired product in comparable yields.
Having established the optimal conditions for the reaction between benzoyl chloride and lithiated 1-bromo-4-methoxybenzene 1, we next evaluated the substrate scope for various acid chlorides with 1-bromo-4-methoxybenzene 1 as the benchmark bromide coupling partner.
In general, aryl acid chlorides with electron donating or electron neutral substituents tolerated our optimised system well affording yields ranging from 62 to 77% (Table 2, entries 1–5). Aromatic acid chlorides with electron withdrawing groups (EWG) afforded the undesired over-addition byproduct in significant amounts (entry 6, 3f, 42%, 4f, 39%) presumably due to strong activation of the ketone to further nucleophilic attack. The use of aliphatic acid chlorides such as hexanoyl chloride and pivaloyl chloride were also tolerated, affording moderate to good yields of ketones (entries 7 and 8, 3g, 55% and 3h, 71%) with low to minimal formation of the tertiary alcohol or tertiary alcohol derived side product.14 Next, 6-chloronicotinoyl chloride was reacted to explore tolerance for heterocyclic substrates. A moderate amount of the desired ketone (entry 9, 3i, 45%) was isolated with significant formation of tertiary alcohol 4i (40%). Improved results were seen with thiophene-2-carbonyl chloride (entry 10) affording the desired 2-thiopene substituted ketone 3j in moderate yield (55%) and a small amount of tertiary alcohol 4j (13%). Use of ethyl 4-(4-methoxybenzoyl)benzoate showed sensitive functionalities such as an ester functional group could be tolerated, affording 3k as the major product in 43% yield with trace amounts of tertiary alcohol.
|
||||
|---|---|---|---|---|
| Entry | Acid chloride | Product (ketone) | Isolated yield (ketone), % | Isolated yield (tert-alchohol), % |
| 1 |  |
 |
66 | 13 4a |
| 2 |  |
 |
63 | 9 4b |
| 3 |  |
 |
62 | <3 4c |
| 4 |  |
 |
77 | <3 4d |
| 5 | 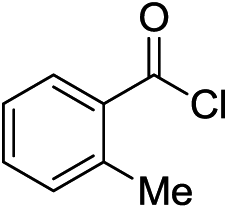 |
 |
74 | 10 4e |
| 6 |  |
 |
42 | 39 4f |
| 7 |  |
 |
55 | <3 4g |
| 8 |  |
 |
71 | <3 4h |
| 9 |  |
 |
45 | 40 4i |
| 10 | 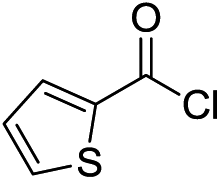 |
 |
55 | 13 4j |
| 11 |  |
 |
43 | <3 4k |
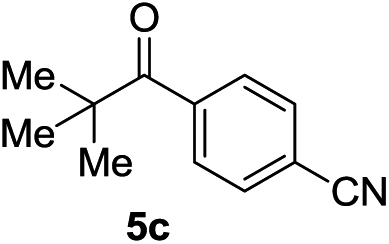
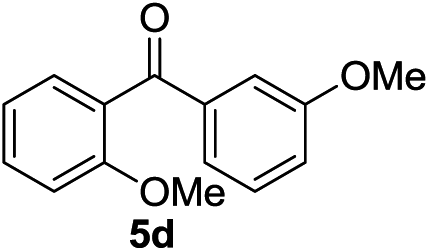
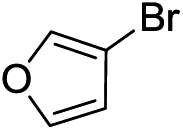
Next, we furthered the scope by introducing variations to both the bromide and acid chloride substrate (Table 3). Aliphatic pivaloyl chloride was well tolerated in the reaction with 4-methylphenyl-, 3-thienyl- and 4-cyanophenylbromides affording moderate to excellent yields of 5a, 5b and 5c (Table 3, entries 1–3, 50–86%). The effects of increased steric hindrance and electron donating character on the aromatic acid chloride coupling partner were investigated in the reaction of 2- or 3-methoxybenzoyl chloride with 3-methoxyphenyl-, 3-furyl- and 3-thienylbromides to afford the desired aromatic and heterocyclic ketones in good yields ranging from 65 to 72% (entries 4–6). Similarly, the reaction of 2-methylbenzoyl chloride with 4-methoxyphenyl- and 3-furylbromides afforded ketones 5g and 5h in moderate to good yields (entries 7–8, 56%, 75%) with only trace amounts of the undesired tertiary alcohols formed.
|
|||||
|---|---|---|---|---|---|
| Entry | Acid chloride | Arl bromide | Product (ketone) | Isolated yield (ketone), % | Isolated yield (tert-alchohol), % |
| 1 |  |
 |
 |
65 | <3 6a |
| 2 |  |
 |
 |
86 | <3 6b |
| 3 |  |
 |
 |
50 | <3 6c |
| 4 |  |
 |
 |
72 | <3 6d |
| 5 | 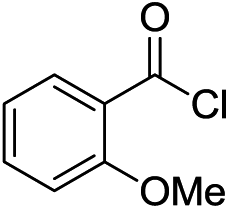 |
 |
 |
65 | <3 6e |
| 6 |  |
 |
 |
68 | <3 6f |
| 7 | 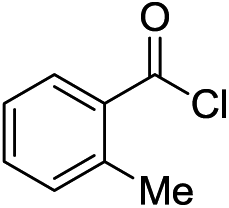 |
 |
 |
56 | <3 6g |
| 8 |  |
 |
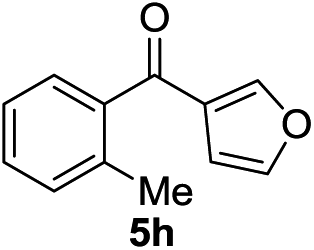 |
75 | <3 6h |

When scaling up traditional chemistry, there are several disadvantageous factors that requires mitigation, such as inefficient heating, mixing, cooling, and accidental runaway of exotherm,15 and in generation of large amounts of waste.
In contrast, a key advantage of a continuous flow system is the inherent simplicity and reliability in the reaction scale up9,16 afforded by precise control of heating, mixing and cooling parameters of chemicals in micro reactors.
To demonstrate the scalability of our method in the synthesis of substituted ketones, the reaction between 3-bromothiophene and pivaloyl chloride was repeated on a 5 mmol scale under our optimised flow conditions to afford the desired heterocyclic ketone 5b in 85% yield (Fig. 3). It should be noted that the total collection and residence times for this particular reaction were 12.5 minutes and 11.2 seconds respectively, rendering our process efficient and effective for scale up operations.
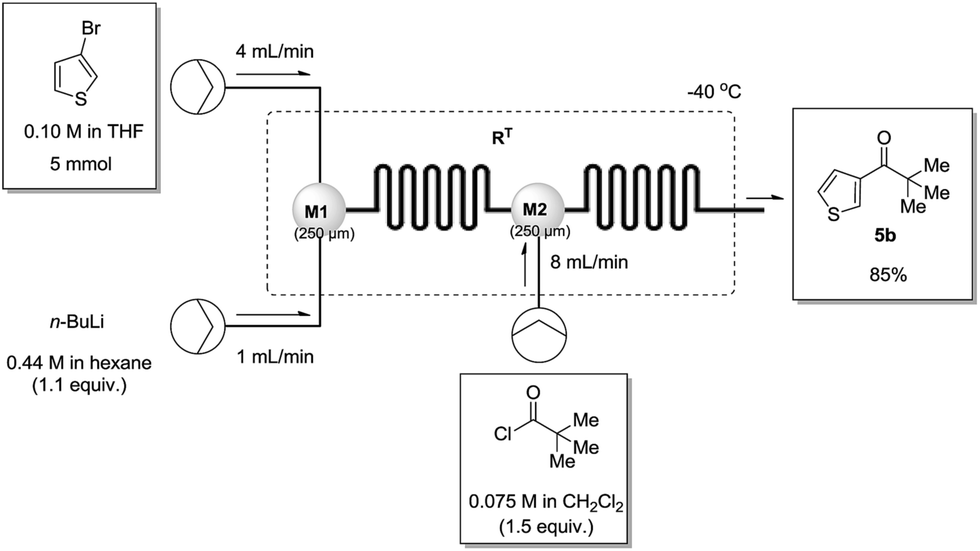 | ||
| Fig. 3 Large scale synthesis of 2,2-dimethyl-1-(thiophen-3-yl)propan-1-one 5b (5 mmol scale) using optimised continuous flow conditions. | ||
In summary, reaction between organolithiums and acid chlorides under continuous flow conditions have been developed to afford substituted ketones with minimal formation of the over-alkylated adducts for most cases. The reaction readily proceeds at −40 °C within a short residence time of only 11.2 seconds, and the scalability of our method has been demonstrated by the successful large scale preparation of ketone 5b. The method presented herein may serve to represent yet another reaction traditionally inaccessible by batch operations but enabled by continuous flow reactors.
Acknowledgements
This work was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF) (2015R1A1A1A05001334) and the New & Renewable Energy of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant (no. 20133030000210).Notes and references
- (a) A. M. Prince, D. Pascual, D. Meruelo, L. Liebes, Y. Mazur, E. Dubovi, M. Mandel and G. Lavie, Photochem. Photobiol., 2000, 71, 188 CrossRef CAS; (b) H. Surburg, J. Paten, Common Fragrance and Flavour Materials, Wiley-VCH, Weinheim, 5th edn, 2006 Search PubMed; For a selected use of ketones in synthesis of natural product see: (c) J. R. Huckins, J. de Vicente and S. D. Rychnovsky, Org. Lett., 2007, 9, 4757 CrossRef CAS PubMed; For selected use of ketones in synthesis of pharmaceutically relevant molecule see: (d) C.-Y. Chen, L. F. Frey, S. Shultz, D. J. Wallace, K. Marcantonio, J. F. Payack, E. Vazquez, S. A. Springfield, G. Zhou, P. Liu, G. R. Kieczykowski, A. M. Chen, B. D. Phenix, U. Singh, J. Strine, B. Izzo and S. W. Krska, Org. Process Res. Dev., 2007, 11, 616 CrossRef CAS.
- (a) P. H. Gore, Chem. Rev., 1955, 55, 229 CrossRef CAS; (b) A. Furstner, D. Voigtlander, W. Schrader, D. Giebel and M. T. Reetz, Org. Lett., 2001, 3, 417 CrossRef CAS; (c) J. Ross and J. Xiao, Green Chem., 2002, 4, 129 RSC; (d) S. Gmouh, H. Yang and M. Vaultier, Org. Lett., 2003, 5, 2219 CrossRef CAS PubMed; (e) E. Fillion, D. Fishlock, A. Wilsily and J. M. Goll, J. Org. Chem., 2005, 70, 1316 CrossRef CAS PubMed.
- (a) C. Malanga, L. A. Aronica and L. Lardicci, Tetrahedron Lett., 1995, 36, 9185 CrossRef CAS; (b) M. Arisawa, Y. Torisawa, M. Kawahara, M. Yamanaka, A. Nishida and M. Nakagawa, J. Org. Chem., 1997, 62, 4327 CrossRef CAS PubMed; (c) R. K. Dieter, Tetrahedron, 1999, 55, 4177 CrossRef CAS; (d) X.-J. Wang, L. Zhang, X. Sun, Y. Xu, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2005, 7, 5593 CrossRef CAS PubMed.
- (a) M. K. Eberle and G. G. Kahle, Tetrahedron Lett., 1980, 21, 2303 CrossRef CAS; (b) G. M. Rubottom and C. W. Kim, J. Org. Chem., 1983, 48, 1550 CrossRef CAS.
- (a) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815 CrossRef CAS; (b) T. Mukaiyama, M. Araki and H. Takei, J. Am. Chem. Soc., 1973, 95, 4763 CrossRef CAS; (c) J. K. Park, W. K. Shin and D. K. An, Tetrahedron Lett., 2013, 54, 3199 CrossRef CAS PubMed; (d) P.-Q. Huang, Y. Wang, K.-J. Xiao and Y.-H. Huang, Tetrahedron, 2015, 71, 4248 CrossRef CAS PubMed.
- (a) H. Gilman and J. M. Straley, Recl. Trav. Chim. Pays-Bas, 1936, 55, 821 CrossRef CAS PubMed; (b) G. M. Whitesides, C. P. Casey, J. San Filippo Jr and E. Panek, Trans. N. Y. Acad. Sci., 1967, 29, 572 CrossRef CAS PubMed; (c) G. H. Posner, C. E. Whitten and P. McFarland, J. Am. Chem. Soc., 1972, 94, 5106 CrossRef CAS; (d) G. H. Posner and C. E. Whitten, Org. Synth., 1976, 55, 122 CrossRef CAS; (e) N. Yoshikai, R. Iida and E. Nakamura, Adv. Synth. Catal., 2008, 350, 1063 CrossRef CAS PubMed.
- (a) D. E. Bergbreiter and J. M. Killough, J. Org. Chem., 1976, 41, 2750 CrossRef CAS; (b) J. W. Labadie and J. K. Stille, J. Am. Chem. Soc., 1983, 105, 669 CrossRef CAS; (c) J. W. Labadie and J. K. Stille, J. Am. Chem. Soc., 1983, 105, 6129 CrossRef CAS; (d) R. A. Grey, J. Org. Chem., 1984, 49, 2288 CrossRef CAS; (e) G. W. Kabalka, R. R. Malladi, D. Tejedoor and S. Kelley, Tetrahedron Lett., 2000, 41, 999 CrossRef CAS; (f) D. Wang and Z. Zhang, Org. Lett., 2003, 5, 4645 CrossRef CAS PubMed; (g) A. H. Cherney and S. E. Reisman, Tetrahedron, 2014, 70, 3259 CrossRef CAS PubMed; (h) L. Axelsson, J.-B. Veron, J. Savmarker, J. Lindh, L. R. Odell and M. Larhed, Tetrahedron Lett., 2014, 55, 2376 CrossRef CAS PubMed; (i) D. Lee, T. Ryu, Y. Park and P. H. Lee, Org. Lett., 2014, 16, 1144 CrossRef CAS PubMed; (j) F. Rafiee and A. R. Hajipour, Appl. Organomet. Chem., 2015, 29, 181 CrossRef CAS PubMed.
- T. M. Bare and H. O. House, Org. Synth., 1969, 49, 81 CrossRef CAS.
- For comprehensive books and treaties on continuous flow chemistry see: (a) Chemical Reactions and Processes under Flow Conditions, ed., S. V. Luis, E. Garcia-Verdugo, RSC Green Chemistry, Cambridge, 2010 Search PubMed; (b) Flow Chemistry: Organic Synthesis in Motion, ed., F. Darvas, V. Hessel, G. Dorman, De Gruyter, Berlin, 2014, vol. 1 Search PubMed; (c) V. Hessel, Design and engineering of microreactor and smart-scaled flow processes, MDPI AG, Basel, 2015 Search PubMed; (d) J. Yoshida, Basics of Flow Microreactor Synthesis, Springer Japan, Tokyo, 2015 Search PubMed.
- For recent reviews on continuous flow chemistry and its application see: (a) S. V. Ley, D. E. Fitzpatrick, R. M. Myers, C. Battilocchio and R. J. Ingham, Angew. Chem., Int. Ed., 2015, 54, 10122 CrossRef CAS PubMed; (b) S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449 CrossRef CAS PubMed; (c) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688 CrossRef CAS PubMed; (d) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194 CrossRef CAS; For recent examples of continuous flow chemistry involved in synthesis of medicinally important molecules see: (e) D. Ghislieri, K. Gilmore and P. H. Seeberger, Angew. Chem., Int. Ed., 2015, 54, 678 CAS; (f) B. Pieber, T. Glasnov and C. O. Kappe, Chem.–Eur. J., 2015, 21, 4368 CrossRef CAS PubMed; (g) S.-H. Lau, A. Galvan, R. R. Merchant, C. Battilocchio, J. A. Souto, M. B. Berry and S. V. Ley, Org. Lett., 2015, 17, 3218 CrossRef CAS PubMed.
- J. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896 RSC.
- J. Wu, X. Yang, Z. He, X. Mao, T. A. Hatton and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 153, 8416 CrossRef PubMed.
- H. Kim, H.-J. Lee and D.-P. Kim, Angew. Chem., Int. Ed., 2014, 54, 1877 CrossRef PubMed.
- In the reaction between 1-bromo-4-methoxybenzene 1 and hexanoyl chloride (Table 2, entry 7), significant amount of olefin 4,4'-(hex-1-ene-1,1-diyl)bis(methoxybenzene) 4g′ was isolated in 16% yield presumably derived from β-elimination of the tertiary alcohol 4g.
- C. Toulouse, J. Cezerac, M. Cabassud, M. V. Le Lann and G. Casamatta, Chem. Eng. Sci., 1996, 51, 2243 CrossRef CAS.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14890a |
| This journal is © The Royal Society of Chemistry 2015 |