
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A self-assembling polycationic nanocarrier that exhibits exceptional gene transfection efficiency†
Kishor
Sarkar
 a,
Sai Rama
Krishna Meka
b,
Giridhar
Madras
a and
Kaushik
Chatterjee
*b
a,
Sai Rama
Krishna Meka
b,
Giridhar
Madras
a and
Kaushik
Chatterjee
*b
aDepartment of Chemical Engineering, Indian Institute of Science, Bangalore 560012, India
bDepartment of Materials Engineering, Indian Institute of Science, Bangalore 560012, India. E-mail: kchatterjee@materials.iisc.ernet.in; Tel: +91-80-2293-3408
First published on 22nd October 2015
Abstract
The lack of an efficient and safe carrier is a major impediment in the field of gene therapy. Although gelatin (GT), a naturally derived polymer, is widely used in drug delivery applications, it is unable to bind DNA efficiently. In this study, a novel polycationic gene carrier was prepared by conjugation of low molecular weight polyethyleneimine (LPEI) with GT through 4-bromonaphthaleic anhydride as a coupling agent to avoid self crosslinking. Self-assembly of LPEI conjugated GT (GT–LPEI) with plasmid DNA (pDNA) yielded nanoparticles with high gene complexation ability to form ∼250 nm cylindrical nanoparticles with a zeta potential of ∼27 mV. GT–LPEI showed exceptionally high transfection efficiency (>90%) in various mammalian cells including primary stem cells with minimal cytotoxicity. The transfection efficiency of GT–LPEI significantly surpassed that of many commercial reagents. The high gene transfection expression was confirmed in vivo. Thus, GT–LPEI is shown to be a promising nonviral carrier for potential use in gene therapy.
1. Introduction
Despite the development of several carriers in recent years, the lack of an efficient and safe carrier remains a key bottleneck in the field of gene therapy.1–3 Owing to several advantages such as low toxicity, nonimmunogenicity, versatility in chemical modification and low production cost, nonviral carriers have attracted tremendous attention over their viral counterparts in the field of gene therapy.4,5 Liposomes and cationic polymers such as polyethyleneimine (PEI), chitosan and dendrimers are the most widely used nonviral carriers but they exhibit low transfection efficiency with high toxicity.6–8 Among the cationic polymers, PEI is considered as the “gold standard” due to its unique advantages including substantial buffering capacity and high transfection efficiency.9 High molecular weight branched PEI (25 kDa) is the most widely used nonviral carrier due to its high transfection efficiency.10 But its severe toxicity limits its application. On the other hand, low molecular weight PEI (2 kDa) induces lower cytotoxicity but with significantly lower transfection efficiency.11Several extracellular barriers such as degradation by serum nucleases, nonspecific delivery and intracellular barriers including lysosomal degradation, cytosolic degradation and poor nuclear transport are the major limiting factors typically affecting nonviral gene transfection.12,13 To overcome these barriers, various strategies such as targeted delivery through ligand conjugation and improvement in cytosolic delivery using fusogenic peptides have been employed to enhance transfection efficiency.14–17 However, the transfection efficiency of nonviral carriers is still much lower than the viral vectors.
Recently, natural polymers such as proteins, peptides and polysaccharides have been widely used for drug delivery applications due their inherent nontoxicity, biodegradability and biocompatibility.18–21 Among these, gelatin (GT) has a long history of successful use as an additive in pharmaceutics, food items and cosmetics. GT is approved as a GRAS (generally regarded as safe) material by the FDA (United States Food and Drug Administration).22 GT obtained from collagen through alkaline or acid hydrolysis is a denatured protein.23 GT possesses recognition sites such as the RGD (Arg-Gly-Asp) sequence to facilitate easy binding to cells.24 GT nanoparticles have been most widely used for drug delivery applications such as antimicrobial,25 anticancer,26 anti-diabetic,27 anti-inflammatory28 and anti-HIV29 drugs among others. However, GT is less studied for gene delivery applications. In the last decade, Amiji and co-workers have reported on GT nanoparticle based gene delivery vehicles for systemic and oral gene therapy.30–32 A few other groups have also used modified GT for gene delivery.33,34
GT has low cationic charge and as a result it has low complexation capability with negatively charged DNA. Modification of GT with low molecular weight PEI (LPEI) offers a potential route to enhance its DNA complexation ability. LPEI is less cytotoxic than its higher molecular weight counterparts although its transfection efficiency is lower due to poor DNA complexation ability. Conjugation of GT to LPEI was envisaged to provide a minimally cytotoxic carrier with cell binding sites and good DNA complexation ability for high transfection efficiency. However, GT contains both carboxylic acid and primary amine groups. Hence, there is a strong tendency to self-crosslink during cationic modification through conjugation of cationic polymers such as PEI using the conventional carbodiimide coupling reaction. It is envisaged that water soluble LPEI conjugated GT (GT–LPEI) can efficiently complex with pDNA by self-assembly into a compact nanoparticle as a vehicle for gene transfection. Thus, the objective of this study was to prepare GT–LPEI without crosslinking of GT and assess its potential for gene delivery.
Herein, we report on the synthesis of water soluble GT–LPEI using 4-bromonaphthaleic anhydride as the coupling agent. The reaction was a two stage process including the reaction between GT and 4-bromonaphthaleic anhydride to block the primary amine groups of GT followed by conjugation of LPEI through substitution reaction of the bromide groups by primary amine groups of LPEI. Self-assembled GT–LPEI/pDNA nanoparticles were prepared by complex coacervation method through electrostatic interactions between the positively charged GT–LPEI and the negatively charged pDNA. The transfection efficiency and cell viability of GT–LPEI were assessed in a variety of mammalian cells and confirmed in vivo. The cellular uptake mechanisms and intracellular distribution were also characterized.
2. Materials and methods
Type-B gelatin (225 bloom strength) with an isoelectric point of 4.7–5.2 and weight average molecular weight (Mw) 47.5 kDa, branched polyethyleneimine (1.8 and 25 kDa), 4-bromo-1,8-naphthaleic anhydride, anhydrous dimethyl sulfoxide, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), ethidium bromide, chlorpromazine (CPZ), genistein (GEN), amiloride (AMIL), nocodazole (NOC), cytocalasin D (Cy-D) and 2,4,6-trinitrobenzene sulfonic acid (TNBS) were purchased from Sigma-Aldrich (USA). SnakeSkin cellulose acetate dialysis tubing (3.5 kDa MW cut off) was obtained from Thermo Scientific. Dulbecco's modified Eagle's medium (DMEM), knockout DMEM, alpha minimum essential medium (α-MEM), penicillin–streptomycin, trypsin and fetal bovine serum (FBS) were purchased from Invitrogen-Gibco (Carlsbad, Calif).Plasmid DNA (pDNA) encoding for green fluorescence (LifeAct-TagGFP2) and luciferase gene (Luc+) (PGL3-Luc pDNA) under the control of the cytomegalovirus promoter/enhancer were obtained from Ibidi (San Diego, CA) and Promega (USA), respectively. D-Luciferin-sodium salt was obtained from BioVision. Vectors were propagated in competent Escherichia coli DH5α cells. Ultrapure endotoxin free plasmid DNA was prepared using the QIAfilter Midi kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Plasmid DNA was diluted in ultrapure water (Invitrogen) to a final concentration of 1 mg mL−1 DNA. All other reagents were analytical grade and were used directly without further modification.
2.1. Synthesis of PEI conjugated gelatin
Low molecular weight PEI conjugated GT (GT–LPEI) was synthesized according to a previous report on preparation of PEI conjugated chitosan.35 The synthesis of GT–LPEI consisted of two consecutive steps (i) synthesis of N-(4-bromonaphthalimide)-gelatin (Comp 3) followed by (ii) conjugation of LPEI with GT through a substitution reaction as shown in Fig. 1. Briefly, 1.0 g GT (Comp 1) was dissolved into 50 mL DMSO containing 4-bromo-1,8-naphthalic anhydride (Comp 2) at different weight ratios as shown in Table 1. The reaction mixture was refluxed at 80 °C under nitrogen atmosphere for 3 h. Thereafter, the reaction mixture was dialyzed using cellulose acetate dialysis membrane against double distilled water for 3 days to remove DMSO and unreacted Comp 2. Finally, the product was lyophilized for 3 days to get a brown colored Comp 3. | ||
| Fig. 1 Schematic diagram for synthesis of GT–LPEI and cellular uptake of GT–LPEI/pDNA complex. | ||
| Sample code | Composition (weight ratio) | % NH2a | Zeta potential (mV) | Molecular weight (Mw) (kDa) | ||
|---|---|---|---|---|---|---|
| GT | Comp 2 | LPEI | ||||
| a Determined by TNBS assay. | ||||||
| GT–LPEI-0 | 1 | 0.00 | 0 | 14.0 ± 3.5 | 2.4 ± 1.9 | 47.5 |
| GT–LPEI-0.01 | 1 | 0.01 | 50 | 17.2 ± 1.6 | 5.2 ± 2.6 | 48.3 |
| GT–LPEI-0.06 | 1 | 0.06 | 50 | 23.4 ± 2.3 | 11.9 ± 4.2 | 50.3 |
| GT–LPEI-0.12 | 1 | 0.12 | 50 | 31.0 ± 3.2 | 18.5 ± 5.1 | 55.2 |
| GT–LPEI-0.18 | 1 | 0.18 | 50 | 36.7 ± 2.7 | 26.4 ± 3.5 | 56.4 |
| GT–LPEI-0.20 | 1 | 0.20 | 50 | 39.1 ± 1.8 | 36.5 ± 2.8 | 59.7 |
GT–LPEI was prepared by reaction between Comp 3 and excess amount of PEI aqueous solution as follows. 100 mg of Comp 3 was dispersed in 50 mL aqueous solution of PEI (10% PEI solution) and the reaction mixture was then refluxed at 80 °C under nitrogen atmosphere for 3 h. The product was dialyzed against double distilled water for 3 days to remove unreacted PEI followed by lyophilization for 3 days to get a yellow colored final product GT–LPEI, Comp 4.
2.2. Characterization
The synthesis of GT–LPEI was characterized by Fourier transform infrared (FTIR) spectroscopy using ATR (attenuated total reflection) FTIR spectrometer (Perkin Elmer Spectrum 100 FTIR). All spectra were carried out within the frequency range of 4000–600 cm−1 for 42 consecutive scans. The proton nuclear magnetic resonance (1H NMR) spectra were determined on a Bruker AV 3000 Supercon NMR system (Germany) at 400 MHz using D2O and DMSO-d6 as solvent. Chemical shifts (δ) were reported in ppm using tetramethylsilane (TMS) as an internal reference. The molecular weight of GT–LPEI was determined by gel permeation chromatography (GPC) using 0.1 M sodium chloride solution as eluent at flow rate of 0.3 mL min−1 by Waters PC2 separation module and Waters 2414 refractive index detector. Tri-nitro benzene sulfonic acid (TNBS) assay was carried out for primary amine quantification of GT–LPEI.362.3. Preparation of polymer/pDNA complexes
Polymer/pDNA complexes were prepared by complex coacervation method according to a previous study.37 Gelatin and different GT–LPEI stock solutions dissolved in double distilled water (ddH2O) were diluted to 1 mg mL−1 concentration using ddH2O. Independently, pDNA stock solution was diluted to 100 μg mL−1 concentration in 25 mM sodium sulfate solution. Thereafter, equal volumes (50 μL) of polymer and pDNA solutions were mixed together at different weight ratios (polymer to pDNA weight ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) and immediately vortexed for 20–30 s followed by incubation at room temperature for 20 min to form the polymer/pDNA complexes.
:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) and immediately vortexed for 20–30 s followed by incubation at room temperature for 20 min to form the polymer/pDNA complexes.
2.4. Characterization of polymer/pDNA complexes
The complexation capability of GT and GT–LPEI with pDNA was characterized by the agarose gel electrophoresis assay. Polymer/pDNA complexes at different weight ratios were freshly prepared as described above prior to the electrophoresis assay. Thereafter, polymer/pDNA complexes containing 0.5 μg pDNA in each weight ratio were loaded in 0.8% agarose gel prepared in TAE buffer (40 mM trisacetate and 1 mM EDTA solution) containing ethidium bromide (10 μg mL−1) as DNA visualizer. The gel was run at 100 V for 40 min and subsequently the gel picture was captured using myECL Imager gel documentation (Thermo Scientific).The DNA complexation capability of GT and GT–LPEI was further confirmed by ethidium bromide (EtBr) assay according to a previous study.38 Briefly, pDNA/EtBr complex was prepared at a molar ratio of 10:1 (pDNA:EtBr molar ratio). Equal volume of GT and GT–LPEI solution with respect to pDNA/EtBr complex were added to the pDNA/EtBr complex at different weight ratio (1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) to the pDNA and incubated for 30 min at 25 °C. Subsequently, the fluorescence intensity with excitation of 510 nm and emission of 605 nm was measured using a microplate reader (Synergy HT, Biotek).
The particle size and zeta potential of GT/pDNA and GT–LPEI/pDNA complexes at different weight ratios were measured by dynamic light scattering (DLS) using Zetasizer Nano ZS (Malvern Instrument, UK) at 37 °C. Before measuring the particle size and zeta potential, the polymer/pDNA complexes were freshly prepared at different weight ratios and diluted to 1.0 mL with filtered ddH2O. For each experiment, three samples were run. The effect of medium (DMEM) and serum containing medium (10% FBS supplemented DMEM) on particle size and zeta potential were also measured.
The particle size and morphology of GT/pDNA and GT–LPEI/pDNA complexes were further observed by atomic force microscopy (AFM, Park systems NX-10) and the images were analyzed by XEI AFM image processing software. Freshly prepared 5 μL of polymer/pDNA complex diluted in ddH2O was deposited on freshly cleaved mica foil and dried at room temperature before imaging. The particles were also characterized by transmission electron microscopy (TEM; JEOL, Model-1011) at an accelerating voltage of 80 kV.
2.5. Cell culture
HeLa (human cervical cancer cells), A549 (human lung adenocarcinoma cells), SVEC (mouse microvascular endothelial cells), MC3T3-E1 (mouse osteoblasts), RAW 264.7 (murine macrophages) and human mesenchymal stem cells (hMSCs) were used for transfection and cytotoxicity studies in vitro. All cell lines were obtained from ATCC. Primary bone marrow derived hMSCs from a 25 year old male donor were obtained from Stempeutics, India. HeLa, A549, SVEC and RAW 264.7 cells were cultured in DMEM medium containing 10% (v/v) FBS, 1% penicillin–streptomycin and 1% of 2 mM L-glutamine in 5% CO2 and 37 °C. MC3T3-E1 cells and hMSCs were cultured in α-MEM and knockout DMEM, respectively, containing 10% serum and antibiotics as above.2.6. In vitro cytotoxicity assay
The toxicity of GT and GT–LPEI at different concentrations (0, 10, 20, 50, 100, 200, 300 and 500 μg mL−1) was optimized in HeLa cells. Branched PEIs (2 and 25 kDa) were used as positive controls. 5 × 103 cells suspended in complete culture medium were seeded in each well of a 96-well plate and cultured for 24 h. The polymer solutions at different concentrations were added to the cells and incubated in the CO2 incubator for 24 h. 5.0 μL of MTT solution (5 mg mL−1) diluted to 100 μL in DMEM medium was added to the treated cell and incubated for 4 h. The MTT solution was replaced by 100 μL DMSO to dissolve the formazan crystals. The absorbance of the solution was measured at 570 nm using a microplate reader (Synergy HT, BioTek instrument) to calculate the cell viability (%) as follows:where, OD570(sample) and OD570(control) were the absorbance values of the polymer treated cells and untreated cells, respectively. Three replicates were used for each measurement.
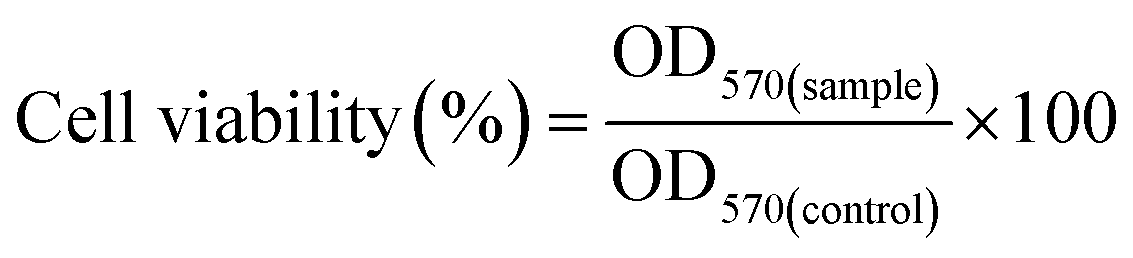
The toxicity of GT/pDNA and GT–LPEI-0.20/pDNA complexes at different weight ratios such as 1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1 was also measured on different cells such as HeLa, A549, SVEC, MC3T3-E1, RAW 264.7 and hMSCs. Freshly prepared polymer/pDNA complexes were added to the cells and incubated for 4 h in CO2 incubator at 37 °C. The polymer/pDNA complex containing solution was replaced by fresh growth medium containing serum and further incubated for 44 h. Lipofectamine 2000 (LF 2000)/pDNA (Life Technologies) and PEI (25 kDa)/pDNA complexes were used as positive controls. The cell viability (%) was measured as described above.
2.7. In vitro transfection study
The transfection efficiency of different GT–LPEIs was optimized in HeLa cells. HeLa cells were seeded in a 24 well plate at a density of 5 × 104 cells per well and cultured until 70–80% confluency. GT/pDNA and GT–LPEI/pDNA complexes were freshly prepared at different weight ratios (1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) as described above containing 1 μg pDNA for each weight ratio. The polymer/pDNA complexes diluted with complete culture medium were added to the cell and incubated for 4 h in CO2 incubator at 37 °C. The transfection medium was replaced by fresh complete culture medium and further incubated for 44 h. pDNA alone and LF 2000/pDNA were used as negative and positive controls, respectively. Branched PEI (25 kDa)/pDNA complex at N/P ratio (nitrogen to phosphate ratio) of 10 was also used as a positive control. The green fluorescence expression of the transfected cells was observed by fluorescence microscopy (Olympus IX53, Japan). Fluorescence-activated cell sorting (FACS, Becton–Dickinson) was used to quantify the transfection efficiency. The transfection efficiency of GT–LPEI-0.20 was further compared with other commercialized transfection reagents such as Lipofectamine 3000 (LF 3000, Life Technologies), DreamFect Gold (DF Gold, OZ Biosciences), TransIT 2020 and TransIT X2 (both Mirus Bio LLC). The effect of serum content on transfection efficiency was also observed.
The transfection efficiency of the carrier was further assessed in different cells such as A549, SVEC, MC3T3-E1 and RAW 264.7 cells and hMSCs. GT–LPEI-0.20/pDNA complexes were freshly prepared at three different weight ratios (20:1, 25:1 and 30:1) and the transfection study performed as above.
2.8. Intracellular distribution and kinetics
To understand the pathways involved in cellular uptake, different endocytic inhibitor drugs such as chlorpromazine (10 μg mL−1), genistein (200 μM) and amiloride (100 μM) were used to block clathrin, caveolae and macropinocytosis mediated endocytosis pathways, respectively. Nocodazole (10 μM), cytochalasin D (20 μM) and RGDS (200 nM) were also used to block cellular cytoskeleton, microtubule and surface integrin, respectively. Low temperature (4 °C) was applied to block the energy-dependent endocytosis. Cells were treated with the different drugs for 1 h prior to the transfection. Thereafter, GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 was added to the drug treated cells in the presence of drugs and incubated for 4 h. The media was replaced by fresh growth media and further incubated for another 20 h. The transfection of untreated cell with GT–LPEI-0.20/pDNA complex was considered as control. The transfection efficiency was quantified by FACS analysis.
In order to track the intracellular distribution of GT–LPEI/DNA complex after cellular uptake, pDNA was fluorescently labeled with Cy3 using Label IT nucleic acid labeling kit (Mirus Bio, USA). HeLa cells were seeded at a density of 1 × 105 cells per well in a 4-well chambered glass slide (SPL Life Sciences) and cultured for 24 h. GT–LPEI-0.20/DNA complex at weight ratio of 25:1 containing 1 μg Cy3-labeled pDNA was added to the cell and incubated for different time points such as 0, 0.5, 2 and 4 h. At each time point, the cells were fixed with 3.7% (w/v) formaldehyde for 30 min and stained with diamidino phenylindole (DAPI) for 10 min. The intracellular distribution of the complexes at different time intervals was observed by confocal laser scanning microscopy (CLSM; Leica TCS Sp5). The intracellular distribution of GT–LPEI-0.20/DNA complex was compared with pDNA alone, PEI (25 kDa)/pDNA complex at N/P ratio of 10 and LF 2000/pDNA complex.
2.9. Lysotracker colocalization assay
GT–LPEI-0.20 and PEI (25 kDa) were labeled with fluorescein isothiocyanate (FITC) according to the previous report.39 1 × 105 HeLa cells were seeded in a 24 well plate and cultured for 24 h. FITC labeled GT–LPEI-0.20/pDNA and PEI (25 kDa)/pDNA complexes at weight ratio of 25:1 and N/P ratio of 10, respectively were prepared prior to the experiment. Polymer/DNA complexes were added to the cells in serum free medium and incubated at 37 °C. After 4 h, the medium containing the polymer/DNA complex was removed and washed thrice with fresh PBS. Lysotracker Red DND 99 (Life Technologies) diluted in serum free medium at concentration of 500 nM was added to the cells followed by incubation for 1 h at 37 °C. Cells were washed three times after removing the medium and subsequently fixed using 4% paraformaldehyde solution followed by counter staining of nuclei by DAPI, and imaged using the CLSM.
2.10. In vivo transfection
To assess transfection in vivo, 6–8 week old, 20–25 g female Balb/c mice were used and the animals were maintained in pathogen free environment at controlled temperature. The animal study was carried out by following the guidelines approved by Institutional Animal Ethics Committee of the Indian Institute of Science. PGL3-Luc pDNA was used for in vivo transfection. 100 μL freshly prepared GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 containing 40 μg pDNA was injected intraperitoneally. pDNA alone and PEI (25 kDa)/pDNA complex at N/P ratio of 10.0 were used as the negative and positive controls, respectively. Three animals (n = 3) were used in each group. The animals were injected intraperitoneally with 200 μL of D-luciferin (15 mg mL−1) 24 h post injection and anesthetized by isoflurane gas (300 mL air flow with 3% isoflurane). The signal of luciferase gene expression was visualized by using bioluminescence IVIS imaging system (Caliper Life Science) after 5 min exposure under continuous supply of isoflurane gas (200 mL air with 2% isoflurane) and the images were captured using the Living Image software.
2.11. Statistical analysis
All the data are shown as the average ± the standard deviation and one-way ANOVA was used to determine statistically significant differences (p values less than 0.05).3. Results and discussion
3.1. Synthesis and characterization of GT–LPEI
GT–LPEI was synthesized by conjugation of low molecular weight PEI (1.8 kDa) with gelatin through naphthalimide moiety as shown in Fig. 1. Gelatin contains both primary amine and carboxylic acid groups. During the conjugation of PEI with gelatin through reaction of the primary amine groups of PEI and the carboxylic acid groups of gelatin by conventional carbodiimide chemistry, there is enhanced tendency to form crosslinked products through reaction of intramolecular primary amine and carboxylic acid groups of gelatin. This may reduce the DNA complexation ability and cellular binding capacity of the modified gelatin. To minimize the formation of crosslinked nanoparticles, the primary amine groups of gelatin were first blocked through reaction with 4-bromonaphthalic anhydride to form Comp 3. Subsequently, PEI was conjugated with Comp 3 through substitution reaction of the primary amine group of PEI and the bromide group of Comp 3 to synthesize GT–LPEI. A library of Comp 3 was synthesized by varying the weight ratios of GT and Comp 2 as shown in Table 1. Excess amount of PEI was reacted with different types of Comp 3 to synthesize different GT–LPEIs.The synthesis of GT–LPEI was characterized by FTIR as shown in Fig. S1.† The FTIR spectra of GT showed a broad peak at 3446 cm−1 corresponding to the overlapped hydrogen bonded O–H (hydroxyl group) stretching with N–H stretching of the primary amine group in GT. The strong absorption peak at 1655 cm−1 for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of amide group indicates the presence of amide linkages in GT. After reaction of GT with Comp 2, two new peaks appeared at 1784 and 1732 cm−1 due to the symmetric and antisymmetric CO stretching of the naphthalimide group, respectively, confirming the synthesis of Comp 3. In addition, the peak for O–H stretching shifted from 3446 cm−1 to 3420 cm−1 and became narrower, which indicates that the primary amine groups of GT reacted with naphthalic anhydride. The O–H stretching peak shifted further to a higher value and became broader after conjugation of PEI with Comp 3 due to the overlap of hydrogen bonded O–H stretching with the N–H stretching of primary amine groups in PEI. The strong absorption peak at 1567 cm−1 due to the N–H stretching of primary amine groups confirms the conjugation of PEI with GT through the naphthalimide moiety.
O stretching of amide group indicates the presence of amide linkages in GT. After reaction of GT with Comp 2, two new peaks appeared at 1784 and 1732 cm−1 due to the symmetric and antisymmetric CO stretching of the naphthalimide group, respectively, confirming the synthesis of Comp 3. In addition, the peak for O–H stretching shifted from 3446 cm−1 to 3420 cm−1 and became narrower, which indicates that the primary amine groups of GT reacted with naphthalic anhydride. The O–H stretching peak shifted further to a higher value and became broader after conjugation of PEI with Comp 3 due to the overlap of hydrogen bonded O–H stretching with the N–H stretching of primary amine groups in PEI. The strong absorption peak at 1567 cm−1 due to the N–H stretching of primary amine groups confirms the conjugation of PEI with GT through the naphthalimide moiety.
The synthesis of GT–LPEI was further validated by 1H NMR spectrometer. The 1H NMR spectra of GT, Comp 3 and GT–LPEI-0.20 are shown in Fig. S2.† The typical 1H NMR spectra of GT (Fig. S2a†) indicates the presence of different amino acid moieties in GT such as valine (Val), leucine (Leu) and isoleucine (Ile) (0.7–0.95 ppm); alanine (Ala) (1.3 ppm); arginine (Arg) (1.6 and 3.1 ppm) and proline (Pro) (3.55 ppm).40 A new broad multiplicity peak appeared at 7.6–8.8 ppm due to the Hα and Hβ protons present in the aromatic ring of the naphthalimide moiety indicating the synthesis of Comp 3 through reaction of GT and Comp 2 (Fig. S2b†). After conjugation of PEI with Comp 3 through substitution reaction of the bromide group of naphthalimide moiety by the primary amine group of PEI, a broad peak with multiplicity appeared at 2.3–3.0 ppm due to –CH2– protons present in PEI confirming the synthesis of GT–LPEI (Fig. S2c†).
From Table 1, it is observed that the molecular weight of GT increased with the increase in the weight ratios of GT and Comp 2 resulting from the conjugation of LPEI with GT through the naphthalimide moiety. GT is insoluble in water at ambient temperature (23 °C) but is soluble in hot water. After reaction of GT with Comp 2, the solubility of GT decreased gradually with increasing weight ratio of GT:Comp 2 (as shown in Table 1) likely due to the increase in the content of hydrophobic aromatic moiety in the product Comp 3. However, all GT derivatives became water soluble at room temperature after conjugation of low molecular weight PEI with Comp 3. During reaction of Comp 3 and PEI, we used excess amount PEI (50 fold excess) to minimize intermolecular crosslinking. When lower amounts of PEI were used, the reaction of one molecule of PEI with two molecule of Comp 3 is more likely yielding water insoluble crosslinked products. In the presence of excess PEI, smaller sized PEI molecule can react preferentially with Comp 3 compared to the larger molecule (Comp 3). Similar phenomenon was observed in a previous study where water soluble chitosan was prepared by conjugation of polyamidoamine dendrimer with chitosan through maleic anhydride.41
3.2. Preparation and characterization of polymer/DNA complexes
A cationic polymer can form complex with negatively charged pDNA through electrostatic interactions. The complexation capability of GT and GT–LPEI was observed by agarose gel electrophoresis assay as shown in Fig. S3.† In agarose gel electrophoresis assay, GT/pDNA and GT–LPEI/PDNA complexes were prepared at different weight ratios (1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) containing 0.5 μg of pDNA in each. From Fig. S3a,† it is seen that GT was not able to complex with all pDNA at any weight ratio due to its low cationic charge or zeta potential (Table 1). The DNA complexation capability of GT significantly increased after conjugation of cationic branched PEI with GT through the naphthalimide moiety. The complexation capability of GT–LPEI also increased with increase in amine content as shown in Table 1 (determined by TNBS assay). All GT–LPEIs of weight ratio of 5:1 and higher were able to complex all of the pDNA.
The DNA complexation capability of GT and GT–LPEI was further observed by EtBr assay as shown in Fig. S4.† In this assay, EtBr molecules intercalate in the base pair of double helix of pDNA resulting in strong fluorescence. As it is a reversible reaction, the addition of the cationic compound can displace the EtBr molecule lowering the fluorescence signal. From Fig. S4,† it can be seen that the relative fluorescence intensity of pDNA/EtBr complex decreased upon addition of GT and GT–LPEI. Due to low cationic charge of GT, the change in relative fluorescence intensity was low. But GT–LPEI sharply decreased the fluorescence intensity due to the large positive charge as shown in Table 1. The fluorescence intensity gradually decreased with the increase in polymer/DNA weight ratio. Among all the different GT–LPEIs, GT–LPEI-0.20 was most efficient due to its highest cationic character (Table 1). Results of the EtBr assay result corroborate the data from the agarose electrophoresis.
The particle size and zeta potential of polymer/DNA complex are important parameters for efficient cellular uptake.42 The particle size and zeta potential of GT/pDNA and GT–LPEI/pDNA complexes were determined by DLS. The average particle size of GT/pDNA and GT–LPEI/pDNA complexes is shown in Fig. 2. Due to inefficient binding capability of GT with pDNA, the GT/pDNA complexes were large and the smallest particle size of GT/pDNA complex was ∼1000 nm at weight ratio of 30:1. But the particle size decreased significantly after conjugation of PEI with GT. GT–LPEI-0.20 formed the smallest particle among the all GT–LPEIs due to its highest amine content and consequently higher zeta potential. The particle size was larger at low weight ratio but it decreased with increase in the weight ratio. GT–LPEI-0.20 formed smallest particle (∼250 nm) with pDNA at weight ratio of 25:1 but the particle size marginally increased with further increase of the weight ratio putatively due to the repulsion of the excessive positive charge offered by GT–LPEI.
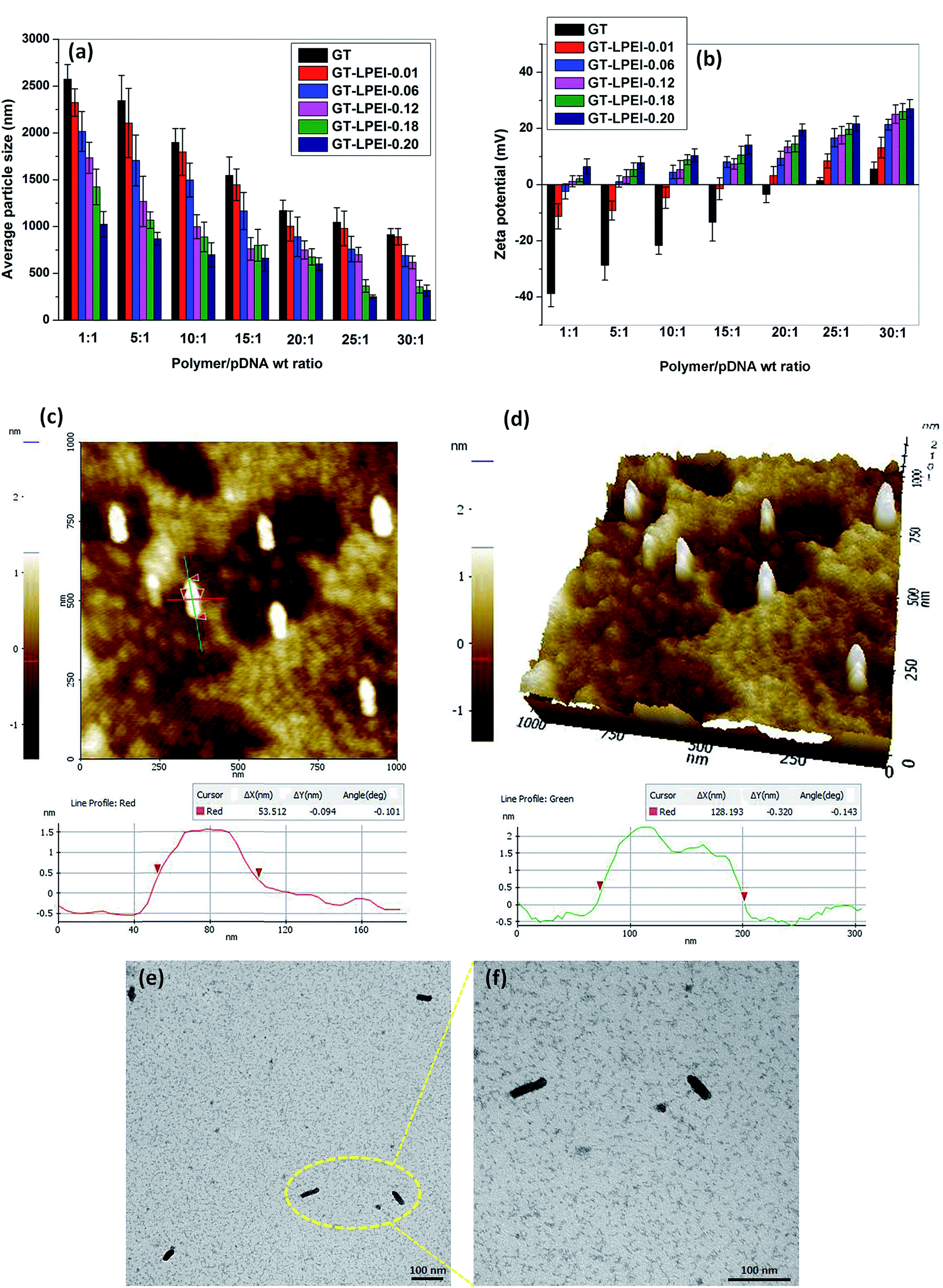 | ||
| Fig. 2 (a) Average particle size and (b) zeta potential of GT/pDNA and GT–LPEIs/pDNA complexes at different weight ratios of 1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1 determined by DLS. AFM images (c) height image and (d) 3D topography images of GT–LPEI-0.20/pDNA complex at weight ratio of 25:1. TEM micrographs of GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 at low magnification (e) and high magnification (f). | ||
Net positive charge of a polymer/DNA complex facilitates cellular uptake through electrostatic interactions between the positively charged complex and the negatively charged cellular membrane. Fig. 2b presents the measured zeta potential of GT/pDNA and GT–LPEI/pDNA complexes at different weight ratios. It can be seen that the zeta potential of GT/pDNA complexes is negative up to weight ratio of 20:1 indicating that GT was not able to neutralize all the negative charges of pDNA because of its low cationic property. In contrast, GT–LPEI/pDNA complexes except GT–LPEI-0.01/pDNA and GT–LPEI-0.06/pDNA complex show positive zeta potential at all weight ratios. The zeta potential of GT–LPEI/pDNA complex increased with the increase in weight ratio. GT–LPEI-0.20/pDNA complex showed highest zeta potential among the all GT–LPEI/pDNA complexes because of its large cationic character. The zeta potential of GT–LPEI-0.20/pDNA complex at weight ratio of 20:1 was 19.5 ± 2.1 mV and did not change markedly at higher ratios. Similar trends in zeta potential were observed in previous studies.43–45 High zeta potential is helpful to stabilize the particle size through electrostatic repulsion. Low zeta potential of GT/pDNA complex may result in formation of large agglomerated particles compared to GT–LPEI/pDNA complexes which possess higher zeta potential.
In addition to the particle size and zeta potential of polymer/DNA complex, the morphology of polymer/DNA complex is also a determining factor for efficient cellular uptake.46 The morphology of GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 was observed by AFM as shown in Fig. 2c and d. The GT–LPEI-0.20/pDNA complex was cylindrical having length of 128 nm and diameter of 53 nm (L/D ratio, aspect ratio of 2.4). TEM images of GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 also showed the cylindrical morphology as shown in Fig. 2e and f. Some studies indicate that cylindrical morphology can facilitate in uptake leading to better transfection efficiency although such as shape dependence is not universal and depends on the carrier. A previous study showed that the particles having cylindrical morphology with aspect ratio of 3 (length 150 nm and diameter 50 nm) showed better cellular uptake compared to spherical particles with aspect ratio 1 (length and diameter 200 nm).47 Another study also reported improved transfection in vivo with elongated particles.48
3.3. In vitro cytotoxicity
Low toxicity along with efficient transfection efficiency of the gene carrier system is a prime requirement for gene therapy applications in vivo. The toxicity of the modified gelatin at different concentrations (0, 10, 20, 50, 100, 200, 300 and 500 μg mL−1) was evaluated in HeLa cells by MTT assay as shown in Fig. S5.† Not surprisingly, GT showed no toxicity in HeLa cell even at high concentration (500 μg mL−1) as also reported previously.49 However, the cell viability decreased after conjugation of PEI with GT although the toxicity of all GT–LPEIs were significantly lower compared to both high (25 kDa) and low (2 kDa) molecular weight PEI. The toxicity of all GT–LPEIs increased with increase in polymer concentration. GT–LPEI-0.20 showed higher toxicity among the all GT–LPEIs likely due to its highest cationic property. Toxicity of cationic polymers is attributed to the destabilization of the cellular membrane through electrostatic interactions between negatively charged cellular membrane and the positively charged polymer.50 However, GT–LPEI-0.20 showed 50% cell viability at very high concentration of 320 μg mL−1, which was also significantly higher than that of both PEIs.The cell viability of polymer/pDNA complexes at different weight ratios (1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) was also observed in various cells such as HeLa (Fig. S6a†), A549 (Fig. S6b†), SVEC (Fig. S6c†), MC3T3-E1 (Fig. S6d†), RAW 264.7 (Fig. S6e†) and hMSCs (Fig. S6f†). Cell viability of GT–LPEI-0.20/pDNA complexes at all weight ratios was 80% or better in all cells. Note that viability appears significantly improved in contrast to that of the uncomplexed polycation (Fig. S5†). The zeta potential remains high for the uncomplexed polycationic carrier but it becomes lower after complexation with the negatively charged DNA through charge neutralization. Bare GT–LPEI-0.20 possesses high zeta potential value of 67.7 ± 3.2 mV but the value is lower (27.4 ± 2.5 mV) after complexation with pDNA at 30:1. Moreover, the actual concentration of polymer in polymer/pDNA complex used was less than 100 μg mL−1 of the polymer which yielded good transfection as discussed below. The toxicity of GT–LPEI-0.20/pDNA complexes increased slightly with the increase in the polymer/DNA weight ratio but the cell viability at the highest weight ratio (30:1) was higher compared to both LF 2000/pDNA and PEI (25 kDa)/pDNA complexes in all cells.
3.4. In vitro transfection
Transfection efficiency of GT–LPEI was optimized in HeLa cells under different conditions. Fluorescence micrographs of cells transfected by GT/pDNA and the different GT–LPEIs/pDNA complexes at weight ratios of 20:1, 25:1 and 30:1 are shown in Fig. S7.† GT/pDNA complexes at all weight ratios did not show any transfection likely due to inefficient binding with pDNA as reported above. Transfection efficiency of GT increased after conjugation of PEI with GT because of the increased DNA binding capacity. GT–LPEI-0.20/pDNA complexes showed the highest transfection efficiency at all weight ratios among all the GT–LPEIs prepared because of its superior properties in terms of size, morphology and DNA binding capability, as discussed above.
The transfection efficiency of all GT–LPEIs was further quantified by FACS analysis as shown in Fig. 3. The transfection efficiency of GT significantly increased after conjugation of PEI with GT. Transfection efficiency of all GT–LPEIs increased with the increase in weight ratio. GT–LPEI-0.20 showed highest transfection efficiency and reached up to ∼97% which was markedly higher than that of PEI (25 kDa)/pDNA complex (efficiency of only ∼30%). The transfection efficiency of GT–LPEI-0.20/pDNA complexes at weight ratios of 20:1, 25:1 and 30:1 was further compared with that of different commercially available transfection reagents including LF 2000, LF 3000, DF Gold, TransIT 2020 and TransIT X2 as shown in Fig. 3b. Strikingly, it was observed that GT–LPEI-0.20 showed superior transfection efficiency at all weight ratios compared to all commercialized transfection reagents. Among the commercialized reagents, LF 3000 showed highest transfection efficiency of ∼65% which was significantly lower compared to that seen with GT–LPEI-0.20.
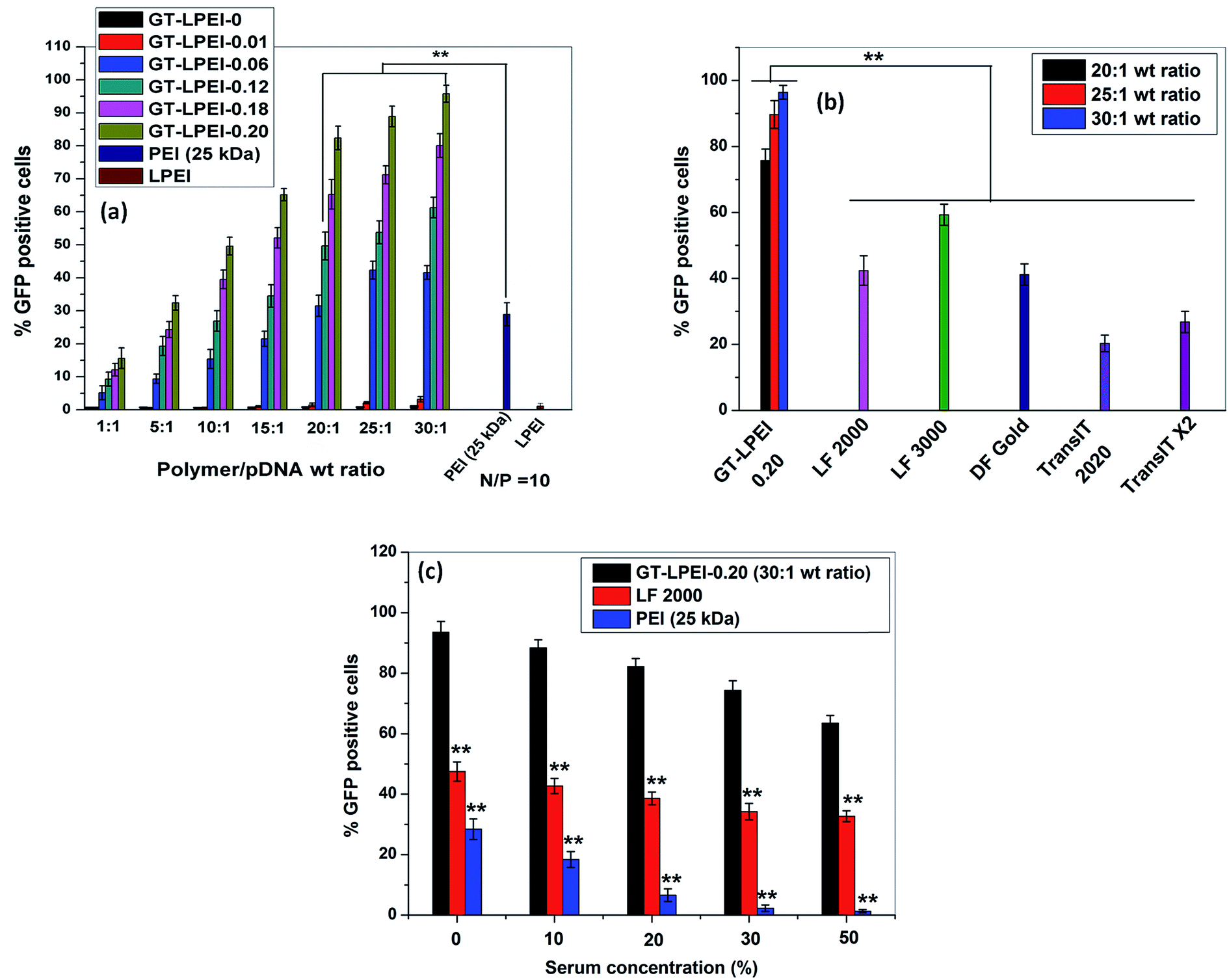 | ||
| Fig. 3 (a) Flow cytometric analysis of GFP-expressing cells 48 h post transfection by GT/pDNA and different GT–LPEIs/pDNA complexes at different weight ratios of 1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1, PEI/pDNA complex at N/P ratio 10 containing 1 μg of pDNA′ in each formulation in HeLa cells, (b) comparative transfection efficiency determined by FACS analysis of GT–LPEI-0.20/pDNA complex at weight ratios 20:1, 25:1 and 30:1 with commercialized transfection reagents and (c) effect of serum concentration on transfection efficiency of GT–LPEI-0.20/pDNA complex at weight ratio of 25:1. LF 2000/pDNA and PEI (25 kDa)/pDNA complexes were used as positive controls. | ||
The transfection efficiency of gene delivery system in presence of serum proteins is critical for successful gene therapy in vivo. Generally, it has been found that the transfection efficiency of a polymeric gene delivery system decreases sharply in the presence of serum proteins due to unwanted interactions between the protein and the gene delivery system.51 The effect of serum proteins on transfection efficiency of GT–LPEI-0.20 was assessed by varying the serum concentration from 0% to 50% as shown in Fig. 3c. The transfection efficiency of GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 decreased with the increase in serum concentration although the efficiency was ∼70% even at 50% serum concentration. In contrast, LF 2000 and PEI (25 kDa) yielded in only ∼35% and <3% transfection, respectively at 50% serum concentration. It was found that the transfection efficiency marginally decreased in medium containing 10% serum compared to serum-free media. The particle size and zeta potential of GT–LPEI-0.20/pDNA complexes at different weight ratio were also measured as shown in Fig. S8† to assess the effect of serum proteins. The particle size increased in presence of serum containing media likely due to adsorption of negatively charged serum proteins through electrostatic interactions. Similarly, the zeta potential became negative at low weight ratio but became positive beyond weight ratio of 15:1 possibly because of the adsorbed proteins.
It was also observed that the morphology of transfected cells was not affected after transfection using GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 as shown in representative micrographs for HeLa cells (Fig. S9†). GT–LPEI-0.20 alone was also used as the control during transfection. It was observed that the cells remained healthy similar to the negative control indicating that the GT–LPEI carrier is not cytotoxic even when transfection was above 90%.
The transfection efficiency of GT–LPEI-0.20/pDNA complex at weight ratio of 20:1, 25:1 and 30:1 was further evaluated in various cells such as A549, SVEC, MC3T3-E1, RAW 264.7 and hMSCs. The fluorescence micrographs of all the transfected cells are shown in Fig. S10.† GT–LPEI-0.20/pDNA complexes at all weight ratios showed discernibly higher transfection compared to LF 2000 in all the cell lines and the primary human stem cells. To visually demonstrate the high transfection efficiency of GT–LPEI/pDNA complex, the nuclei of the all cells were stained by DAPI and overlaid as merged micrographs as shown in Fig. 4. Note that nearly all the cells in the different cell types were indeed transfected as there was good overlap between GFP expression and nuclear staining. The cells appeared healthy and well spread with no apparent signs of cytotoxicity. The transfection efficiency was further quantified by FACS analysis to validate the fluorescence and staining data. Fig. 5a shows the transfection efficiency of GT–LPEI-0.20/pDNA complex at weight ratios of 20:1, 25:1 and 30:1 quantified by FACS in the various cells. The transfection efficiency of GT–LPEI-0.20 at all weight ratios was significantly higher compared to that of LF 2000 and PEI (25 kDa). The exceptionally high transfection efficiency of GT–LPEI-0.20 may be attributed to its ability to large DNA binding capacity by self assembly, cylindrical morphology and the large availability of cell-adhesive peptide sequences such as RGD in the gelatin which likely augmented cellular uptake and intracellular transport.
 | ||
| Fig. 4 Representative fluorescence micrographs of different GFP transfected and nuclear stained cells by GT–LPEI-0.20/pDNA complexes at weight ratio of 25:1 containing 1 μg of pDNA. The scale bar is 10 mm. | ||
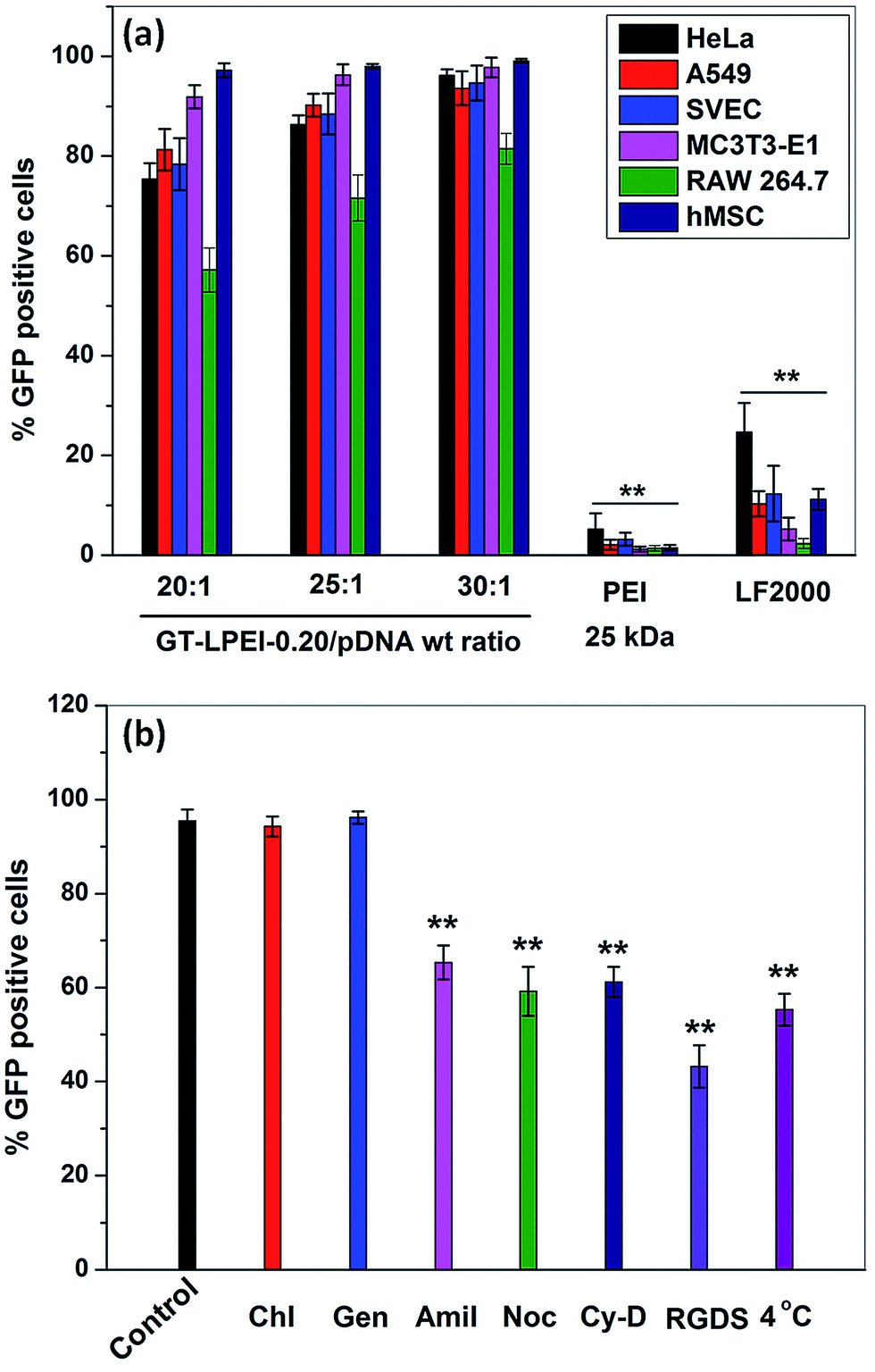 | ||
| Fig. 5 (a) Flow cytometric analysis of GFP-expressing cells 48 h post transfection by GT/pDNA and GT–LPEI-0.20/pDNA complexes at different weight ratios of 20:1, 25:1 and 30:1, PEI/pDNA complex at N/P ratio 10 and LF 2000/pDNA complex containing 1 μg of pDNA in each formulation against various cells and (b) relative transfection efficiency determined by FACS analysis of GT–LPEI-0.20/pDNA complex at the weight ratio of 25:1 against HeLa cell after treating with various endocytotic inhibitors for 1 h before transfection containing 1 μg of pDNA followed by further incubation for 48 h. | ||
3.5. Intracellular kinetics
Intracellular uptake mechanism is another important parameter that determines gene transfection efficiency along with other parameters such as DNA complexation capability, particle size, morphology and zeta potential.52,53 Clathrin, caveolae and macropinocytosis mediated endocytosis pathways are the major uptake pathways for cellular internalization of a polymer/DNA complex.54 To understand the uptake mechanism, the transfection efficiency of GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 was assessed in HeLa cells in the presence of different drugs, peptides and low temperature conditions such as chlorpromazine, genistein, amiloride, nocodazole, cytochalasin D, exogenous soluble RGDS peptide and 4 °C temperature. Fig. 5b shows that low temperature significantly decreased the transfection efficiency of GT–LPEI-0.20 indicating that the cellular uptake of GT–LPEI-0.20/pDNA complex occurred through energy dependent endocytosis pathway. The transfection efficiency was not affected by chlorpromazine and genistein demonstrating that clathrin and caveolae mediated endocytosis pathway were not involved in the cellular uptake of the GT–LPEI-0.20/pDNA complex. In contrast, amiloride significantly inhibited the transfection efficiency (∼32% inhibition) indicating that macropinocytosis mediated endocytosis was the primary uptake mechanism of the GT–LPEI-0.20/pDNA complex. A previous study reported that the clathrin mediated endocytosis pathway is primarily involved for particles of size <200 nm but caveolae and macro-pinocytosis mediated endocytosis are the primary pathways for particles >200 nm.55,56 In this study, the particle size of GT–LPEI-0.20/pDNA complex was around 250 nm which are in good agreement with the previous report. Khalil et al.57 reported that the macropinocytosis mediated endocytosis pathway is more favorable for gene transfection efficiency than clathrin endocytosis because macropinocytosis does not involve degradative lysosome compartment during internalization.
The transgene expression is not only dependent on cellular uptake pathway but also depends on the rate of endosomal release inside the cell.58 El-Sayed et al.59 reported the effect of surface modification of nanoparticle by octaarginine (R8) and octalysine (K8) on endosomal escape rate and consequently the transfection efficiency. Both R8 and K8 modified nanoparticles entered into the cell through macropinocytosis mediated pathway but R8 modified nanoparticle showed better transfection efficiency. They explained that R8 modified nanoparticle showed higher escape rate from endosome due to the positive charge. In this report, GT–LPEI-0.20/pDNA complex with positive zeta potential at weight ratio of 25:1 also followed the macropinocytosis mediated endocytosis pathway. Therefore, GT–LPEI-0.20/pDNA complex might have higher endosomal escape rate resulting in high transfection efficiency. It is also observed from Fig. 5b, the transfection efficiency was also inhibited by nocodazole and cytochalasin D demonstrating that the nuclear transport of GT–LPEI-0.20/pDNA complex occurred through actin and microtubule. Previous reports suggest that the nuclear internalization of polymer/DNA through actin and microtubule enhances the transfection efficiency.60,61 The transfection efficiency was also reduced by the presence of exogenous RGDS peptide indicating that integrin mediated cellular uptake is involved during uptake of the complex.
3.6. Intracellular distribution and kinetics
Intracellular distribution of polymer/DNA complex after cellular uptake is an important parameter for efficient transgene expression. After cellular uptake, the polymer/DNA complex has to overcome several obstacles in the form of transport and degradative mechanisms within the cell. To understand the intracellular distribution kinetics of GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 using Cy3 labeled pDNA, the CLSM images were captured at different cellular uptake time points (0, 0.5, 2 and 4 h) as shown in Fig. S11.† It is observed that a few complexes (red dots) entered into the cell 0.5 h post transfection and the number of red dots (complexes) increased with time. Few complexes reached the nucleus within 2 h post transfection and the number of complexes at the nucleus further increased at 4 h demonstrating rapid nuclear transport of the complexes which facilitates high transfection efficiency.
The intracellular distribution of GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 was also compared to PEI (25 kDa)/pDNA and LF 2000/pDNA complexes as shown in Fig. S12.† pDNA alone was used as the negative control. It can be seen that few PEI (25 kDa)/pDNA complexes entered into the cell after 4 h post transfection and most of the complexes were located away from the nucleus suggesting poor intracellular transport leading to low transfection efficiency of PEI (25 kDa). In contrast, the number of complexes associated with both LF 2000/pDNA and GT–LPEI-0.20/pDNA complex was higher but GT–LPEI-0.20/pDNA complexes showed fastest transport and augmented transfection efficiency.
Cellular internalization after uptake is a major internal barrier in nonviral gene delivery systems.62 It has been reported that most nonviral vectors enter cells through clathrin mediated endocytosis pathway followed by internalization in to degradative lysosomal compartment resulting in low transfection efficiency.63 Lysotracker colocalization assay was performed to assess the involvement of lysosomal compartment in the uptake and intracellular transport of the GT–LPEI carrier (Fig. 6). It was observed that most of the PEI (green) colocalized with the lysosomes (red) as indicated by the arrows (yellow spots) demonstrating that PEI/pDNA complexes are entrapped in the lysosomal degradative compartment resulting in lower transfection efficiency. In contrast, the GT–LPEI-0.20/pDNA complex escaped the lysosomal compartment (minimal colocalization) that likely results in higher transfection efficiency.
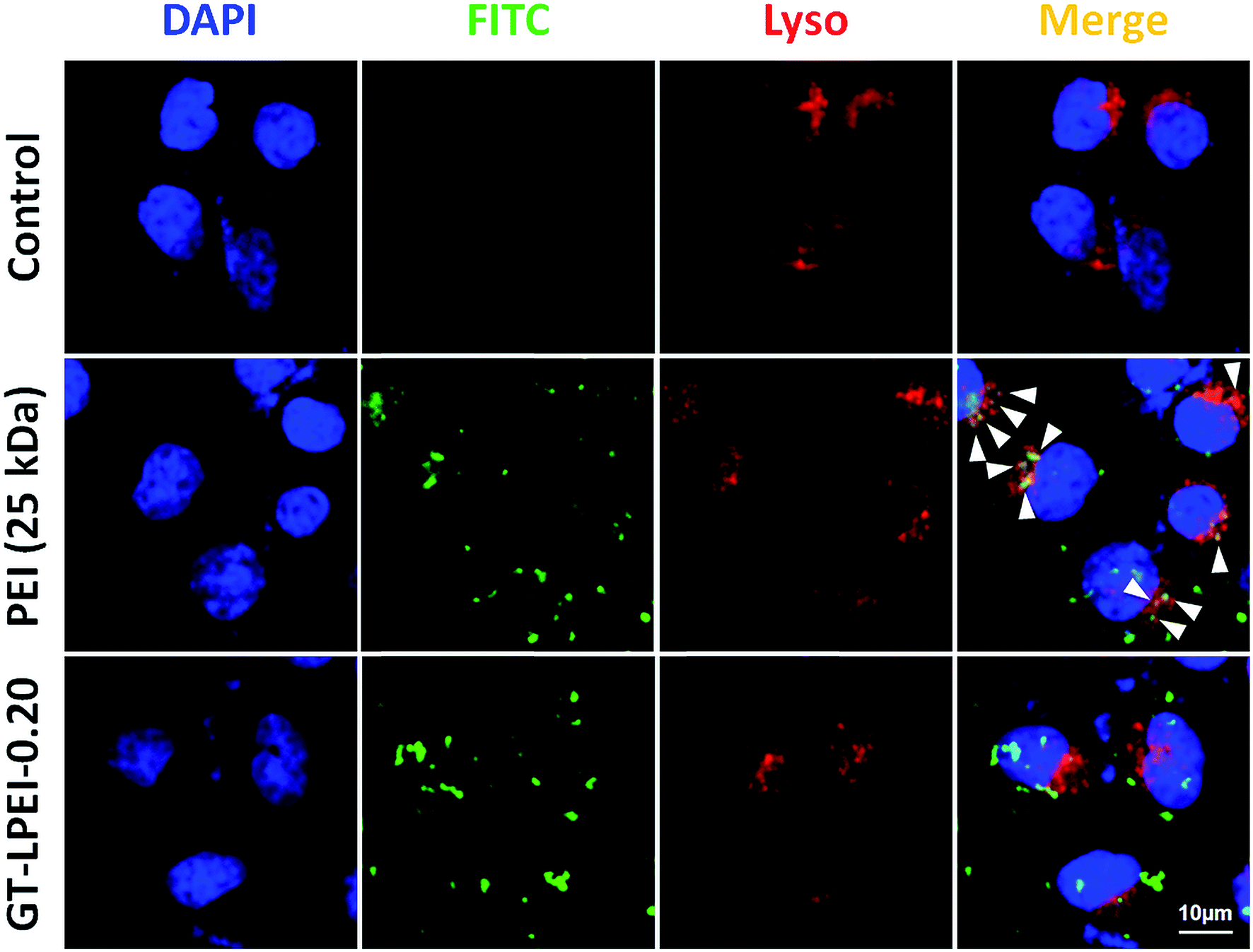 | ||
| Fig. 6 CLSM images of Lysotracker colocalization assay in HeLa cells after 4 h of transfection with FITC-labelled PEI/pDNA complex at N/P ratio of 10 and GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 containing 1 μg pDNA in each formulation. For each panel, images from left to right show stained cell nuclei (blue), FITC labelled polymer (green), Lysotracker (red) and colocalization of green and red in merged (yellow). Scale bar is 10 μm. | ||
3.7. In vivo transfection
For clinical gene therapy application, the gene delivery system should be able to delivery therapeutic gene efficiently and safely in vivo. Toward development for clinical use, a preliminary experiment was performed in mice to assess the efficiency of GT–LPEI-0.20/pDNA complex injected intraperitoneally. pDNA alone and PEI (25 kDa)/pDNA complexes were used as the negative and positive controls, respectively. The luciferase expression was visualized 24 h post injection as shown in representative images in Fig. 7. Strong signal from the peritoneum of the animal injected with GT–LPEI-0.20/pDNA complex indicating the GT–LPEI-0.20 is able to transfect in vivo. In contrast, pDNA and PEI (25 kDa)/pDNA did not show any measurable gene expression also supports the in vitro data. Therefore, the novel GT–LPEI complex developed herein is a promising nonviral carrier for gene therapy that may be further developed for clinical use.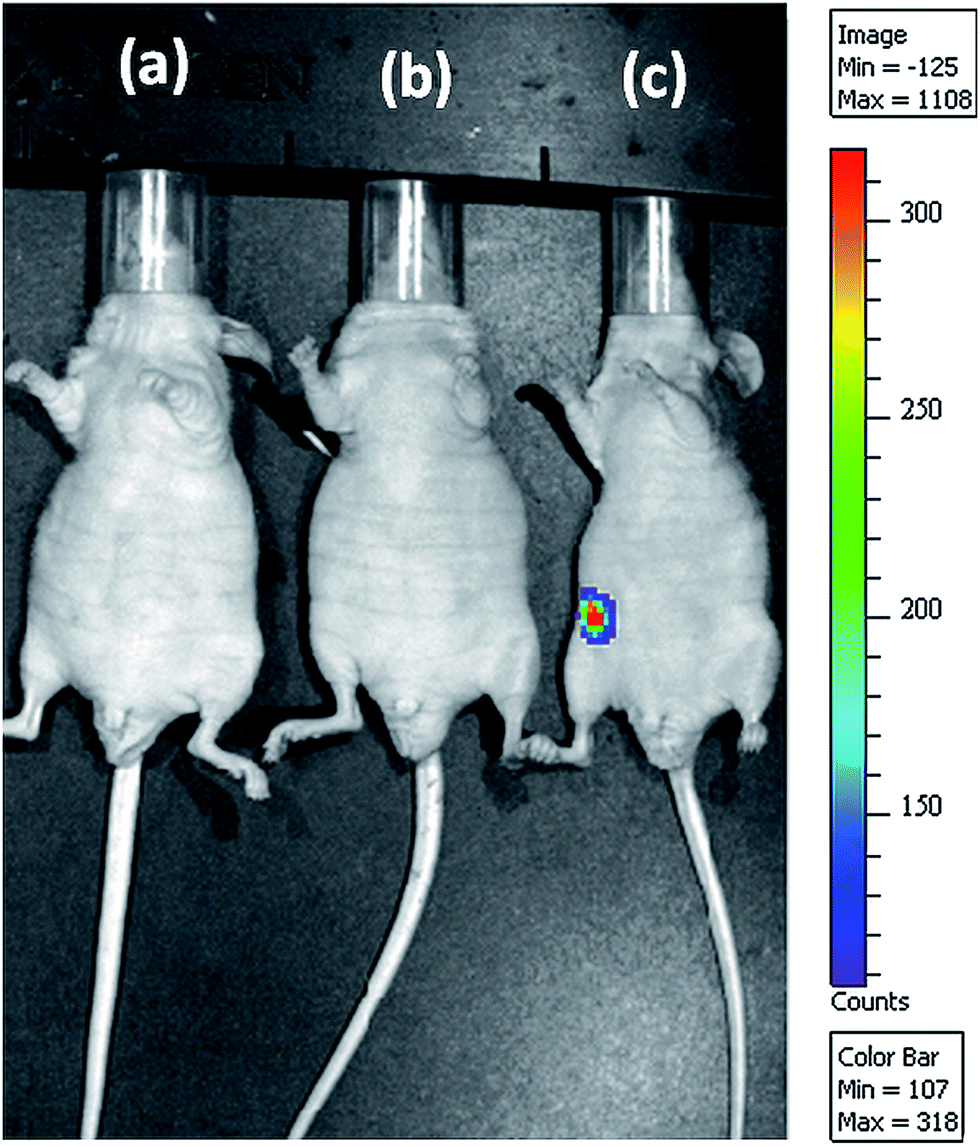 | ||
| Fig. 7 Representative bioluminescence live animal imaging of gene expression 24 h after intraperitoneal injection of (a) luciferase-encoding pDNA only, (b) PEI (25 kDa)/pDNA complex at N/P ratio of 10 and (c) GT–LPEI-0.20/pDNA complex at weight ratio of 25:1 containing 40 μg pDNA in each formulation. The scale indicates surface radiance (photons per s per cm2 per steradian). | ||
4. Conclusion
LPEI was conjugated with gelatin through naphthalimide moiety to avoid self-crosslinking of gelatin to yield GT–LPEI. The LPEI content was optimized to yield GT–LPEI-0.20 with good pDNA complexation ability. Self-assembly of GT–LPEI-0.20 with pDNA yielded cylindrical nanoparticles of ∼250 nm in size with zeta potential of ∼27 mV. GT–LPEI-0.20 showed exceptionally high transfection in a wide variety of mammalian cells surpassing the efficiency offered by many different commercially available transfection reagents. GT–LPEI uptake is shown to be mediated by macro-pinocytosis followed by rapid transport to the nucleus facilitated by F-actin with microtubules without entrapment in the lysosomal compartment. The efficacy of GT–LPEI system in vivo was also demonstrated. Therefore, GT–LPEI is a promising nonviral carrier for gene therapy.Financial disclosure
Patent application has been filed.Acknowledgements
This work was funded by the Nanomission Program of the Department of Science and Technology (DST), India. K. S. was supported by the D. S. Kothari fellowship (BSR/EN/13-14/0005) from the University Grants Commission (UGC), India. K. C. acknowledges the Ramanujan fellowship from DST. The authors thank AFMM, the NMR Center and the Central Confocal Imaging Facility for access to equipments.References
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- M. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671–681 CrossRef CAS PubMed.
- P. P. Kundu and K. Sarkar, in Biopolymers: Biomedical and Environmental Applications, ed. S. Kalia and L. Avérous, John Wiley & Sons, Inc., 2011, DOI:10.1002/9781118164792.ch20, pp. 575–603.
- X. Guo and L. Huang, Acc. Chem. Res., 2011, 45, 971–979 CrossRef PubMed.
- K. Ewert, A. Zidovska, A. Ahmad, N. Bouxsein, H. Evans, C. McAllister, C. Samuel and C. Safinya, in Nucleic Acid Transfection, ed. W. Bielke and C. Erbacher, Springer, Berlin Heidelberg, 2010, vol. 296, ch. 70, pp. 191–226 Search PubMed.
- S. C. de Smedt, J. Demeester and W. E. Hennink, Pharm. Res., 2000, 17, 113–126 CrossRef CAS.
- K. Sarkar, R. Srivastava, U. Chatterji and P. Kundu, J. Appl. Polym. Sci., 2011, 121, 2239–2249 CrossRef CAS.
- U. Lungwitz, M. Breunig, T. Blunk and A. Göpferich, Eur. J. Pharm. Biopharm., 2005, 60, 247–266 CrossRef CAS PubMed.
- M. Thomas and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14640–14645 CrossRef CAS PubMed.
- K. Kunath, A. von Harpe, D. Fischer, H. Petersen, U. Bickel, K. Voigt and T. Kissel, J. Controlled Release, 2003, 89, 113–125 CrossRef CAS PubMed.
- C. H. Jones, C.-K. Chen, A. Ravikrishnan, S. Rane and B. A. Pfeifer, Mol. Pharmaceutics, 2013, 10, 4082–4098 CrossRef CAS PubMed.
- D. Lechardeur and G. L. Lukacs, Curr. Gene Ther., 2002, 2, 183–194 CrossRef CAS PubMed.
- K. Kunath, T. Merdan, O. Hegener, H. Häberlein and T. Kissel, J. Gene Med., 2003, 5, 588–599 CrossRef CAS PubMed.
- S. Mahor, B. C. Dash, S. O'Connor and A. Pandit, Bioconjugate Chem., 2012, 23, 1138–1148 CrossRef CAS PubMed.
- T. Merdan, K. Kunath, H. Petersen, U. Bakowsky, K. H. Voigt, J. Kopecek and T. Kissel, Bioconjugate Chem., 2005, 16, 785–792 CrossRef CAS PubMed.
- V. B. Morris, C. K. S. Pillai and C. P. Sharma, Polym. Int., 2011, 60, 1097–1106 CrossRef CAS.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- M. Bachar, A. Mandelbaum, I. Portnaya, H. Perlstein, S. Even-Chen, Y. Barenholz and D. Danino, J. Controlled Release, 2012, 160, 164–171 CrossRef CAS PubMed.
- J. Kundu, Y.-I. Chung, Y. H. Kim, G. Tae and S. Kundu, Int. J. Pharm., 2010, 388, 242–250 CrossRef CAS PubMed.
- N. Saranya, A. Moorthi, S. Saravanan, M. P. Devi and N. Selvamurugan, Int. J. Biol. Macromol., 2011, 48, 234–238 CrossRef CAS PubMed.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 161, 38–49 CrossRef CAS PubMed.
- P. Kozlov and G. Burdygina, Polymer, 1983, 24, 651–666 CrossRef CAS.
- H. Wang, O. C. Boerman, K. Sariibrahimoglu, Y. Li, J. A. Jansen and S. C. Leeuwenburgh, Biomaterials, 2012, 33, 8695–8703 CrossRef CAS PubMed.
- M. Nahar, D. Mishra, V. Dubey and N. K. Jain, Nanomedicine: Nanotechnology, Biology and Medicine, 2008, 4, 252–261 CrossRef CAS PubMed.
- E. Leo, M. A. Vandelli, R. Cameroni and F. Forni, Int. J. Pharm., 1997, 155, 75–82 CrossRef CAS.
- Y.-Z. Zhao, X. Li, C.-T. Lu, Y.-Y. Xu, H.-F. Lv, D.-D. Dai, L. Zhang, C.-Z. Sun, W. Yang and X.-K. Li, Acta Diabetol., 2012, 49, 315–325 CrossRef CAS PubMed.
- R. Kumar, R. C. Nagarwal, M. Dhanawat and J. K. Pandit, J. Biomed. Nanotechnol., 2011, 7, 325–333 CrossRef CAS PubMed.
- S. K. Jain, Y. Gupta, A. Jain, A. R. Saxena, P. Khare and A. Jain, Nanomedicine: Nanotechnology, Biology and Medicine, 2008, 4, 41–48 CrossRef CAS PubMed.
- G. Kaul and M. Amiji, J. Pharm. Sci., 2005, 94, 184–198 CrossRef CAS PubMed.
- M. D. Bhavsar and M. M. Amiji, AAPS PharmSciTech, 2008, 9, 288–294 CrossRef CAS PubMed.
- M. Bhavsar and M. Amiji, Gene Ther., 2008, 15, 1200–1209 CrossRef CAS PubMed.
- G. K. Zorzi, J. E. Párraga, B. Seijo and A. Sánchez, Macromol. Biosci., 2011, 11, 905–913 CrossRef CAS PubMed.
- X.-H. Tian, Z.-G. Wang, H. Meng, Y.-H. Wang, W. Feng, F. Wei, Z.-C. Huang, X.-N. Lin and L. Ren, Int. J. Nanomed., 2013, 8, 865 CrossRef PubMed.
- K. Sarkar, M. Debnath and P. Kundu, Carbohydr. Polym., 2013, 92, 2048–2057 CrossRef CAS PubMed.
- C. N. Grover, J. H. Gwynne, N. Pugh, S. Hamaia, R. W. Farndale, S. M. Best and R. E. Cameron, Acta Biomater., 2012, 8, 3080–3090 CrossRef CAS PubMed.
- K. Sarkar, A. Chatterjee, G. Chakraborti and P. P. Kundu, Carbohydr. Polym., 2013, 98, 596–606 CrossRef CAS PubMed.
- K. Sarkar, S. L. Banerjee, P. P. Kundu, G. Madras and K. Chatterjee, J. Mater. Chem. B, 2015, 3, 5266–5276 RSC.
- Q. Hu, J. Wang, J. Shen, M. Liu, X. Jin, G. Tang and P. K. Chu, Biomaterials, 2012, 33, 1135–1145 CrossRef CAS PubMed.
- X. Hu, D. Li, F. Zhou and C. Gao, Acta Biomater., 2011, 7, 1618–1626 CrossRef CAS PubMed.
- K. Sarkar and P. P. Kundu, Int. J. Biol. Macromol., 2012, 51, 859–867 CrossRef CAS PubMed.
- R. I. Mahato and S. W. Kim, Pharmaceutical Perspectives of Nucleic Acid-Based Therapy, CRC Press, 2003 Search PubMed.
- K. Sarkar and P. Kundu, Carbohydr. Polym., 2013, 98, 495–504 CrossRef CAS PubMed.
- X. Zhao, Z. Li, H. Pan, W. Liu, M. Lv, F. Leung and W. W. Lu, Acta Biomater., 2013, 9, 6694–6703 CrossRef CAS PubMed.
- B. Lu, D.-Q. Wu, H. Zheng, C.-Y. Quan, X.-Z. Zhang and R.-X. Zhuo, Mol. BioSyst., 2010, 6, 2529–2538 RSC.
- J. Shi, J. L. Choi, B. Chou, R. N. Johnson, J. G. Schellinger and S. H. Pun, ACS Nano, 2013, 7, 10612–10620 CrossRef CAS PubMed.
- S. E. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- B. Sternberg, K. Hong, W. Zheng and D. Papahadjopoulos, Biochim. Biophys. Acta Biomembr., 1998, 1375, 23–35 CrossRef CAS.
- B. Gaihre, M. S. Khil and H. Y. Kim, J. Microencapsulation, 2011, 28, 286–293 CrossRef CAS PubMed.
- S. Hong, P. R. Leroueil, E. K. Janus, J. L. Peters, M.-M. Kober, M. T. Islam, B. G. Orr, J. R. Baker and M. M. Banaszak Holl, Bioconjugate Chem., 2006, 17, 728–734 CrossRef CAS PubMed.
- P. P. Kundu and V. Sharma, Curr. Opin. Solid State Mater. Sci., 2008, 12, 89–102 CrossRef CAS.
- M. van der Aa, U. Huth, S. Häfele, R. Schubert, R. Oosting, E. Mastrobattista, W. Hennink, R. Peschka-Süss, G. Koning and D. Crommelin, Pharm. Res., 2007, 24, 1590–1598 CrossRef CAS PubMed.
- C. W. Pouton and L. W. Seymour, Adv. Drug Delivery Rev., 1998, 34, 3–19 CrossRef CAS PubMed.
- A. F. Adler and K. W. Leong, Nano Today, 2010, 5, 553–569 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- A. El-Sayed and H. Harashima, Mol. Ther., 2013, 21, 1118–1130 CrossRef CAS PubMed.
- I. A. Khalil, K. Kogure, S. Futaki and H. Harashima, J. Biol. Chem., 2006, 281, 3544–3551 CrossRef CAS PubMed.
- M. Magzoub, A. Pramanik and A. Gräslund, Biochemistry, 2005, 44, 14890–14897 CrossRef CAS PubMed.
- A. El-Sayed, I. A. Khalil, K. Kogure, S. Futaki and H. Harashima, J. Biol. Chem., 2008, 283, 23450–23461 CrossRef CAS PubMed.
- K. Tanaka, T. Kanazawa, K. Sugawara, S. Horiuchi, Y. Takashima and H. Okada, Int. J. Pharm., 2011, 419, 231–234 CrossRef CAS PubMed.
- L. Pigeon, C. Gonçalves, D. Gosset, C. Pichon and P. Midoux, Small, 2013, 9, 3845–3851 CrossRef CAS PubMed.
- D. Cross and J. K. Burmester, Clin. Med. Res., 2006, 4, 218–227 CrossRef CAS PubMed.
- R. Wattiaux, N. Laurent, S. Wattiaux-de Coninck and M. Jadot, Adv. Drug Delivery Rev., 2000, 41, 201–208 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14829d |
| This journal is © The Royal Society of Chemistry 2015 |