
Highly enantioselective direct allylic alkylation of butenolides with Morita–Baylis–Hillman carbonates catalyzed by chiral squaramide-phosphine†
Tian-Chen Kang,
Xuan Zhao,
Feng Sha* and
Xin-Yan Wu*
Key Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: xinyanwu@ecust.edu.cn; Tel: +86 21 64252011
First published on 24th August 2015
Abstract
An efficient asymmetric vinylogous allylic alkylation of β,γ-butenolides with Morita–Baylis–Hillman carbonates has been developed. With a chiral cyclohexane-based squaramide-phosphine catalyst 5e, optically active γ,γ-disubstituted butenolides containing adjacent quaternary and tertiary chiral centers have been constructed in good-to-excellent yields (up to 98%) and excellent stereoselectivities (87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13–99:1 dr, 96–99% ee).
:13–99:1 dr, 96–99% ee).
β,γ-Unsaturated butenolides have attracted great attention recently due to their utility for the direct construction of γ,γ-disubstituted butenolide derivatives, which are important motifs in biologically active natural compounds and pharmaceutically useful molecules.1–3 Many endeavors have been devoted towards developing efficient protocols to access optically active γ,γ-disubstituted butenolides in the past decade.4 In 2010, Chen and co-workers4a have developed the first direct asymmetric allylic alkylation of γ-substituted β,γ-butenolides with Morita–Baylis–Hillman (MBH) carbonates to access γ,γ-disubstituted butenolides containing adjacent quaternary and tertiary chiral centers, catalyzed by cinchona alkaloid derivatives. Since Chen's pioneering work, various direct vinylogous reactions with β,γ-butenolides as the nucleophiles have been reported, and the electrophiles involve α,β-unsaturated carbonyl compounds,4b,f–k nitroolefins,4e,l aldimines4c and MBH carbonates.4a,d It is worth noting that the progress has been mainly focused on Michael reactions. To the best of our knowledge, enantioselective vinylogous allylic alkylation has been rarely described.4a,d,5 Therefore, the development of a simplified protocol to allow easy access to diverse γ,γ-disubstituted butenolides from β,γ-unsaturated butenolides via vinylogous allylic alkylation is still challenging.
Recently, the enantioselective allylic alkylation with MBH adducts catalyzed by Lewis basic tertiary amines or phosphines has emerged as a powerful strategy to deliver multifunctional compounds.6 Among these examples, the asymmetric vinylogous reaction involving MBH carbonates catalyzed by tertiary phosphines has not been well developed.7 To the best of our knowledge, the only example is the reaction between 2-trimethylsilyloxy furan and MBH carbonates reported by Shi's group (Scheme 1).5a Despite their ability to render γ,γ-stereogenic quaternary centers, which are challenging motifs in organic synthesis, the application of γ-substituted β,γ-butenolides in the asymmetric allylic alkylation is still in its infancy4a,d probably due to the low reactivity. Considering the higher nucleophilicity of tertiary phosphines than the corresponding amines, we envisioned that chiral tertiary phosphines could be more effective for the direct vinylogous allylic alkylation between γ-substituted butenolides and MBH carbonates. Herein, we report the first direct asymmetric vinylogous allylic alkylation of butenolides with MBH carbonates to access γ,γ-disubstituted butenolides using chiral tertiary phosphine as organocatalyst.8
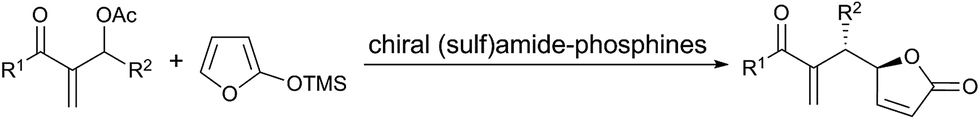 | ||
| Scheme 1 Previous work with chiral phosphine catalysts. | ||
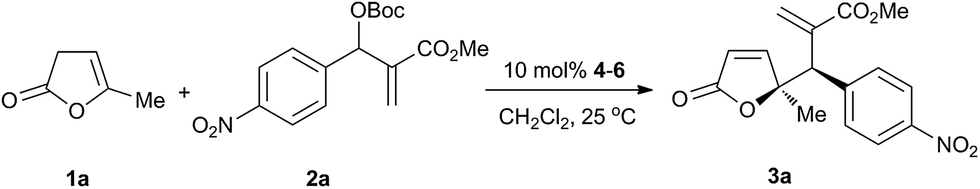
We began our investigation with the vinylogous allylic alkylation of β,γ-butenolide 1a with MBH carbonate 2a in the presence of 10 mol% rac-4a in CH2Cl2 at ambient temperature. To our delight, the vinylogous reaction completed in 4 hours and the regioselective γ,γ-disubstituted butenolide was obtained with excellent diastereoselectivity in 92% yield. Then we examined the chiral cyclohexane-based bifunctional phosphine organocatalysts at 25 °C (Fig. 1), and the results are summarized in Table 1.9 In general, the aliphatic thioureas provided higher yields than the aromatic thioureas with a similar diastereoselectivity (entries 1–4 vs. entries 5 and 6). The bifunctional phosphines with an additional chiral group could enhance the enantioselectivity, and a chirality match existed between the chiral backbone and the additional group (entries 3 and 4). To improve the stereoselectivity, chiral phosphines with other H-bonding donators were examined. Squaramide 5c gave better enantioselectivity but lower yield than the corresponding thiourea 4c (entry 9 vs. entry 3). Therefore squaramide-phosphines10 containing different scaffolds were evaluated (entries 7–11). Organocatalyst 5e bearing aromatic scaffold exhibited better yield and enantioselectivity than 5b and 5c bearing aliphatic scaffold. Using 6a with NH2 group as the H-bonding donor, the vinylogous reaction rate was increased, but poor enantioselectivity obtained. Amide-phosphine 6b with a single H-bonding donator exhibited a lower reactivity, albeit producing the same level of enantioselectivity as the squaramide-phosphines (entry 13). Therefore squaramide-phosphine 5e was selected as the chiral organocatalyst to further optimize the reaction conditions.
 | ||
| Fig. 1 Structures of the chiral bifunctional phosphines screened. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb (%) | drc | eec (%) |
| a The reactions were performed with 0.3 mmol of 1a, 0.2 mmol of 2a, and 10 mol% of catalyst in 1 mL CH2Cl2 at 25 °C.b Isolated yield.c The dr and ee values were based on chiral HPLC analysis, and the given ee data were for 3a.d Not determined.e No reaction. | |||||
| 1 | 4a (R = Bn) | 4 | 94 | 97:3 |
59 |
| 2 | 4b (R = n-C12H25) | 24 | 72 | 97:3 |
73 |
| 3 | 4c (R = (S)-1-phenylethyl) | 12 | 93 | 97:3 |
82 |
| 4 | 4d (R = (R)-1-phenylethyl) | 12 | 52 | 97:3 |
60 |
| 5 | 4e (R = Ph) | 144 | 27 | 97:3 |
82 |
| 6 | 4f (R = 3,5-(CF3)2C6H3) | 24 | Complex | ndd | nd |
| 7 | 5a (R = Bn) | 24 | NRe | nd | nd |
| 8 | 5b (R = n-C12H25) | 3 | 97 | 97:3 |
97 |
| 9 | 5c (R = (S)-1-phenylethyl) | 36 | 84 | 97:3 |
90 |
| 10 | 5d (R = Ph) | 48 | 31 | 97:3 |
95 |
| 11 | 5e (R = 3,5-(CF3)2C6H3) | 12 | 97 | 98:2 |
98 |
| 12 | 6a (R = H) | 3 | 83 | 96:4 |
44 |
| 13 | 6b (R = Bz) | 72 | 85 | 91:9 |
92 |
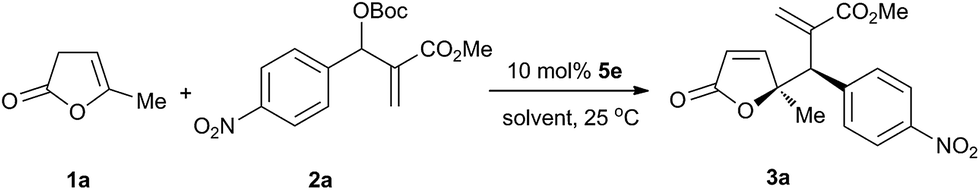
Subsequently, the optimization of other reaction parameters (solvent, ratio of substrates, temperature and substrate concentration) was carried out with squaramide-phosphine 5e. As shown in Table 2, in halogenated solvents γ,γ-disubstituted butenolides 3a could be obtained in excellent yield, diastereoselectivity and enantioselectivity (entries 1–3). Lower yields were produced with other polar solvents surveyed due to the low conversion of the substrates (entries 4–6). Change of the ratio of 1a/2a from 2:1 to 1:1.5 led to higher reaction rate with a similar yield, but the enantioselectivity was slightly lower (entries 2 and 7–9). It was found that higher reaction temperature resulted in an increase of reaction rate with a decrease of diastereoselectivity, without compromising the enantioselectivity (entries 2, 10 and 11). Change of substrate concentration in CHCl3 did not affect the yield and diastereoselectivity. Meanwhile, a slightly higher enantioselectivity was achieved by proceeding the reaction in a lower concentration (entries 2 and 12–15). Further decrease of the concentration from 0.1 M to 0.025 M had no significant impact on stereoselectivity. The decrease of catalyst loading from 10 mol% to 5 mol% led to a significant decrease in yield due to a lower conversion (entry 2 vs. entry 16). Therefore, the optimal reaction conditions have been identified as follows: 1.5 equiv. of β,γ-butenolide 1 and MBH carbonate 2 in CHCl3 (0.1 M) at 25 °C using 10 mol% of squaramide-phosphine 5e as the chiral catalyst.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | Ratio (1a/2a) | Conc. (M) | Time (h) | Yieldb (%) | drc | eec (%) |
| a Unless otherwise noted the reactions were performed with 1a, 2a, and 10 mol% of 5e in the solvent at 25 °C.b Isolated yield.c The dr and ee values were based on chiral HPLC analysis, and the given ee data were for 3a.d Not determined.e At 40 °C.f At 0 °C.g The catalyst loading was 5 mol%. | |||||||
| 1 | CH2Cl2 | 1.5:1 |
0.2 | 12 | 97 | 98:2 |
98 |
| 2 | CHCl3 | 1.5:1 |
0.2 | 12 | 98 | 98:2 |
98 |
| 3 | ClCH2CH2Cl | 1.5:1 |
0.2 | 12 | 92 | 97:3 |
98 |
| 4 | THF | 1.5:1 |
0.2 | 96 | 52 | 29:1 |
96 |
| 5 | EtOAc | 1.5:1 |
0.2 | 96 | 57 | 34:1 |
97 |
| 6 | CH3CN | 1.5:1 |
0.2 | 96 | Trace | ndd | nd |
| 7 | CHCl3 | 1:1.5 |
0.2 | 3 | 98 | 98:2 |
96 |
| 8 | CHCl3 | 1:1 |
0.2 | 9 | 95 | 98:2 |
98 |
| 9 | CHCl3 | 2:1 |
0.2 | 24 | 97 | 98:2 |
98 |
| 10e | CHCl3 | 1.5:1 |
0.2 | 4 | 98 | 97:3 |
98 |
| 11f | CHCl3 | 1.5:1 |
0.2 | 48 | 98 | 99:1 |
98 |
| 12 | CHCl3 | 1.5:1 |
0.4 | 12 | 98 | 98:2 |
97 |
| 13 | CHCl3 | 1.5:1 |
0.1 | 12 | 98 | 98:2 |
99 |
| 14 | CHCl3 | 1.5:1 |
0.05 | 12 | 98 | 98:2 |
99 |
| 15 | CHCl3 | 1.5:1 |
0.025 | 12 | 98 | 98:2 |
99 |
| 16g | CHCl3 | 1.5:1 |
0.2 | 96 | 37 | 98:2 |
99 |
Under the optimized reaction condition, the substrate scope of the vinylogous allylic alkylation using a simple γ-methyl-substituted butenolide 1a and differently substituted MBH carbonates was examined (Table 3, entries 1–16). Excellent enantioselectivities were observed for MBH carbonates derived from different alkyl acrylates and methyl vinyl ketone, while n-butyl acrylate and t-butyl acrylate provided lower yields probably due to the steric effect (entries 1–5). MBH carbonates with a strong electron-withdrawing group at the 4- and 3-position of aromatic ring gave better yields than those with a weak electron-withdrawing group or without substituent (entries 1 and 6–14). MBH carbonates bearing two electron-withdrawing groups at both 3- and 4-positions of phenyl also afforded the desired products in good yields and enantioselectivities (entries 15 and 16). The MBH carbonate with substituent at the 2-position of phenyl failed to produce the desired product probably due to an ortho effect. To our disappointment, MBH carbonates with an electron-donating group such as methyl and methoxyl at phenyl ring, and MBH carbonates of alkyl aldehydes are unreactive substrates under the typical reaction condition.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Time (h) | Yieldb (%) | drc | eec (%) |
| a Unless otherwise noted, the reactions were performed with 0.3 mmol of 1, 0.2 mmol of 2, 10 mol% of 5e in 2 mL CHCl3 at 25 °C.b Isolated yield.c The dr and ee values were based on chiral HPLC analysis, and the given ee data were for 3.d The catalyst loading was 20 mol%.e With 0.2 mmol of 1 and 0.3 mmol of 2. | |||||||
| 1 | Me | 4-NO2 | OMe | 12 | 3a, 98 | 98:2 |
99 |
| 2 | Me | 4-NO2 | OEt | 12 | 3b, 92 | 98:2 |
99 |
| 3 | Me | 4-NO2 | OnBu | 120 | 3c, 56 | 99:1 |
99 |
| 4 | Me | 4-NO2 | OtBu | 120 | 3d, 52 | 99:1 |
99 |
| 5 | Me | 4-NO2 | Me | 6 | 3e, 95 | 99:1 |
99 |
| 6 | Me | 3-NO2 | OMe | 12 | 3f, 98 | 87:13 |
98 |
| 7 | Me | 4-CN | OMe | 12 | 3g, 91 | 97:3 |
98 |
| 8 | Me | 3-CN | OMe | 12 | 3h, 93 | 90:10 |
98 |
| 9 | Me | 4-CF3 | OMe | 72 | 3i, 94 | 96:4 |
98 |
| 10 | Me | 3-CF3 | OMe | 72 | 3j, 87 | 90:10 |
97 |
| 11 | Me | 4-Br | OMe | 120 | 3k, 72 | 96:4 |
98 |
| 12 | Me | 4-Cl | OMe | 96 | 3l, 81 | 96:4 |
98 |
| 13 | Me | 4-F | OMe | 120 | 3m, 74 | 97:3 |
97 |
| 14d | Me | H | OMe | 120 | 3n, 67 | 96:4 |
96 |
| 15 | Me | 3,4-Cl2 | OMe | 12 | 3o, 92 | 94:6 |
98 |
| 16 | Me | 3,4-F2 | OMe | 48 | 3p, 91 | 94:6 |
98 |
| 17 | C6H5 | 4-NO2 | OMe | 14 | 3q, 94 | 88:12 |
99 |
| 18e | C6H5 | 4-NO2 | OMe | 3 | 3q, 94 | 88:12 |
99 |
| 19e | 4-FC6H4 | 4-NO2 | OMe | 3 | 3r, 97 | 93:7 |
98 |
| 20e | 4-ClC6H4 | 4-NO2 | OMe | 6 | 3s, 78 | 98:2 |
98 |
| 21e | 4-MeC6H4 | 4-NO2 | OMe | 2 | 3t, 98 | 91:9 |
99 |
| 22e | 4-MeOC6H4 | 4-NO2 | OMe | 3 | 3u, 94 | 94:6 |
98 |

Further exploration of the substrate scope has been focused on the β,γ-butenolides bearing different γ-aryl groups. Considering the reaction rate, excess MBH carbonate (1.5 equiv.) was used, and the vinylogous reaction was accomplished within 3 hours attaining the same enantiocontrol (entry 18 vs. entry 17). To our delight, the reaction tolerated all the β,γ-butenolides bearing different γ-aryl groups with either an electron-withdrawing or an electron-donating group, affording the corresponding adducts 3 in excellent yields, diastereo- and enantioselectivities (Table 3, entries 17–22).
To determine the absolute configuration, a vinylogous allylic alkylation was performed with DHQD(PYR)2 as chiral catalyst, according to the literature report.4a Compound 3p was assigned as (R,S) by comparing the HPLC spectra and optical rotation values with the product resulting from DHQD(PYR)2 catalysis. The absolute configurations of other products were assigned by analogy.
In conclusion, we have developed the first chiral phosphine-organocatalyzed enantioselective vinylogous allylic alkylation of MBH carbonates with β,γ-butenolides. In the presence of 10 mol% chiral cyclohexane-based squaramide-phosphine 5e, the corresponding γ,γ-disubstituted butenolides containing adjacent quaternary and tertiary stereogenic centers were obtained in up to 98% yield with excellent diastereo- and enantioselectivities. This asymmetric reaction provides an efficient approach toward the synthesis of chiral γ,γ-disubstituted butenolides.
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21242007, 21102043), Science and Technology Commission of Shanghai Municipality (15ZR1409200), and the Fundamental Research Funds for the Central Universities.Notes and references
- For recent reviews of natural products containing butenolides, see: (a) A. D. Rodríguez, Tetrahedron, 1995, 51, 4571 CrossRef; (b) A. Bermejo, B. Figadère, M.-C. Zafra-Polo, I. Barrachina, E. Estornell and D. Cortes, Nat. Prod. Rep., 2005, 22, 269 RSC; (c) J. D. Sellars and P. G. Steel, Eur. J. Org. Chem., 2007, 3815 CrossRef CAS PubMed; (d) T. Montagnon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001 CrossRef CAS PubMed; (e) I. Prassas and E. P. Diamandis, Nat. Rev. Drug Discovery, 2008, 7, 926 CrossRef CAS PubMed; (f) P. A. Roethle and D. Trauner, Nat. Prod. Rep., 2008, 25, 298 RSC; (g) Y. Igarashi, H. Ogura, K. Furihata, N. Oku, C. Indananda and A. Thamchaipenet, J. Nat. Prod., 2011, 74, 670 CrossRef CAS PubMed.
- For some examples on building γ-substituted butenolides with 2-silyloxyfurans, see: (a) C. W. Jefford, D. Jaggi and J. Boukouvalas, Tetrahedron Lett., 1987, 28, 4037 CrossRef CAS; (b) H. Kitajima, K. Ito and T. Katsuki, Tetrahedron, 1997, 53, 17015 CrossRef CAS; (c) H. Kitajima and T. Katsuki, Synlett, 1997, 568 CrossRef CAS; (d) H. Nishikori, K. Ito and T. Katsuki, Tetrahedron: Asymmetry, 1998, 9, 1165 CrossRef CAS; (e) G. Desimoni, G. Faita, S. Filippone, M. Mella, M. G. Zampori and M. Zema, Tetrahedron, 2001, 57, 10203 CrossRef CAS; (f) S. P. Brown, N. C. Goodwin and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 1192 CrossRef CAS PubMed; (g) H. Suga, T. Kitamura, A. Kakehi and T. Baba, Chem. Commun., 2004, 1414 RSC; (h) Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS PubMed; (i) H. Yang and S. Kim, Synlett, 2008, 555 CAS; (j) Q. Zhang, X. Xiao, L. Lin, X. Liu and X. Feng, Org. Biomol. Chem., 2011, 9, 5748 RSC; (k) X. Jusseau, P. Retailleau, L. Chabaud and C. Guillou, J. Org. Chem., 2013, 78, 2289 CrossRef CAS PubMed.
- For examples on building γ-substituted butenolides with 2(5H)-furanones, see: (a) B. M. Trost and J. Hitce, J. Am. Chem. Soc., 2009, 131, 4572 CrossRef CAS PubMed; (b) J. Wang, C. Qi, Z. Ge, T. Cheng and R. Li, Chem. Commun., 2010, 46, 2124 RSC; (c) R. P. Singh, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2010, 132, 9558 CrossRef CAS PubMed; (d) H. Huang, F. Yu, Z. Jin, W. Li, W. Wu, X. Liang and J. Ye, Chem. Commun., 2010, 46, 5957 RSC; (e) Y. Zhang, C. Yu, Y. Ji and W. Wang, Chem.–Asian J., 2010, 5, 1303 CAS; (f) J. Luo, H. Wang, X. Han, L.-W. Xu, J. Kwiatkowski, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 1861 CrossRef CAS PubMed; (g) G. Hou, J. Yu, C. Yu, G. Wu and Z. Miao, Adv. Synth. Catal., 2013, 355, 589 CAS; (h) S. Nakamura, R. Yamaji and M. Hayashi, Chem.–Eur. J., 2015, 21, 9615 CrossRef CAS PubMed.
- For asymmetric vinylogous-type reaction of β,γ-butenolides, see: (a) H.-L. Cui, J.-R. Huang, J. Lei, Z.-F. Wang, S. Chen, L. Wu and Y.-C. Chen, Org. Lett., 2010, 12, 720 CrossRef CAS PubMed; (b) A. Quintard, A. Lefranc and A. Alexakis, Org. Lett., 2011, 13, 1540 CrossRef CAS PubMed; (c) L. Zhou, L. Lin, J. Ji, M. Xie, X. Liu and X. Feng, Org. Lett., 2011, 13, 3056 CrossRef CAS PubMed; (d) X. Huang, J. Peng, L. Dong and Y.-C. Chen, Chem. Commun., 2012, 48, 2439 RSC; (e) M. S. Manna, V. Kumar and S. Mukherjee, Chem. Commun., 2012, 48, 5193 RSC; (f) M. S. Manna and S. Mukherjee, Chem.–Eur. J., 2012, 18, 15277 CrossRef CAS PubMed; (g) W. Zhang, D. Tan, R. Lee, G. Tong, W. Chen, B. Qi, K.-W. Huang, C.-H. Tan and Z. Jiang, Angew. Chem., Int. Ed., 2012, 51, 10069 CrossRef CAS PubMed; (h) V. Kumar and S. Mukherjee, Chem. Commun., 2013, 49, 11203 RSC; (i) D. Yang, L. Wang, D. Zhao, F. Han, B. Zhang and R. Wang, Chem.–Eur. J., 2013, 19, 4691 CrossRef CAS PubMed; (j) U. Das, Y.-R. Chen, Y.-L. Tsai and W. Lin, Chem.–Eur. J., 2013, 19, 7713 CrossRef CAS PubMed; (k) J. Ji, L. Lin, L. Zhou, Y. Zhang, Y. Liu, X. Liu and X. Feng, Adv. Synth. Catal., 2013, 355, 2764 CrossRef CAS PubMed; (l) T. Sekikawa, T. Kitaguchi, H. Kitaura, T. Minami and Y. Hatanaka, Org. Lett., 2015, 17, 3026 CrossRef CAS PubMed.
- For asymmetric vinylogous alkylation of MBH carbonates, see: (a) Y.-Q. Jiang, Y.-L. Shi and M. Shi, J. Am. Chem. Soc., 2008, 130, 7202 CrossRef CAS PubMed; (b) H.-L. Cui, J. Peng, X. Feng, W. Du, K. Jiang and Y.-C. Chen, Chem.–Eur. J., 2009, 15, 1574 CrossRef CAS PubMed; (c) J. Peng, X. Huang, H.-L. Cui and Y.-C. Chen, Org. Lett., 2010, 12, 4260 CrossRef CAS PubMed; (d) L. Jiang, Q. Lei, X. Huang, H.-L. Cui, X. Zhou and Y.-C. Chen, Chem.–Eur. J., 2011, 17, 9489 CrossRef CAS PubMed.
- For recent reviews of organocatalytic asymmetric transformations of modified Morita–Baylis–Hillman adducts, see: (a) T.-Y. Liu, M. Xie and Y.-C. Chen, Chem. Soc. Rev., 2012, 41, 4101 RSC; (b) R. Rios, Catal. Sci. Technol., 2012, 2, 267 RSC.
- For the first phosphine-catalyzed allylic alkylation of butenolides and MBH carbonates, see: C.-W. Cho and M. J. Krische, Angew. Chem., Int. Ed., 2004, 43, 6689 CrossRef CAS PubMed.
- For recent reviews on phosphine organocatalysis, see: (a) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed; (b) S.-X. Wang, X. Han, F. Zhong, Y. Wang and Y. Lu, Synlett, 2011, 2766 CAS; (c) Q.-Y. Zhao, Z. Lian, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 1724 RSC; (d) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588 RSC; (e) L.-W. Xu, ChemCatChem, 2013, 5, 2775 CrossRef CAS PubMed; (f) Y. Xiao, Z. Sun, H. Guo and O. Kwon, Beilstein J. Org. Chem., 2014, 10, 2089 CrossRef PubMed.
- For chiral cyclohexane-based bifunctional phosphines, see: (a) Y.-Q. Fang and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 5660 CrossRef CAS PubMed; (b) K. Yuan, L. Zhang, H.-L. Song, Y. Hu and X.-Y. Wu, Tetrahedron Lett., 2008, 49, 6262 CrossRef CAS PubMed; (c) T. Mita and E. N. Jacobsen, Synlett, 2009, 1680 CAS; (d) K. Yuan, H.-L. Song, Y. Hu and X.-Y. Wu, Tetrahedron, 2009, 65, 8185 CrossRef CAS PubMed; (e) K. Yuan, H.-L. Song, Y. Hu, J.-F. Fang and X.-Y. Wu, Tetrahedron: Asymmetry, 2010, 21, 903 CrossRef CAS PubMed; (f) H.-L. Song, K. Yuan and X.-Y. Wu, Chem. Commun., 2011, 47, 1012 RSC; (g) C.-C. Wang and X.-Y. Wu, Tetrahedron, 2011, 67, 2974 CrossRef CAS PubMed; (h) J.-Y. Qian, C.-C. Wang, F. Sha and X.-Y. Wu, RSC Adv., 2012, 2, 6042 RSC; (i) W. Yang, F. Sha, X. Zhang, K. Yuan and X. Wu, Chin. J. Chem., 2012, 30, 2652 CAS; (j) X. Zhao, T.-Z. Li, J.-Y. Qian, F. Sha and X.-Y. Wu, Org. Biomol. Chem., 2014, 12, 8072 RSC; (k) Y.-Q. Fang, P. M. Tadross and E. N. Jacobsen, J. Am. Chem. Soc., 2014, 136, 17966 CrossRef CAS PubMed; (l) X. Zhao, J.-J. Gong, K. Yuan, F. Sha and X.-Y. Wu, Tetrahedron Lett., 2015, 56, 2526 CrossRef CAS PubMed; (m) G. Wang, R. Rexiti, F. Sha and X.-Y. Wu, Tetrahedron, 2015, 71, 4255 CrossRef CAS PubMed.
- For reviews of chiral squaramides in enantioselective organocatalysis, see: (a) J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem.–Eur. J., 2011, 17, 6890 CrossRef PubMed; (b) R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra and HPLC chromatograms of the products. See DOI: 10.1039/c5ra14667d |
| This journal is © The Royal Society of Chemistry 2015 |