
Improved oral bioavailability of docetaxel by nanostructured lipid carriers: in vitro characteristics, in vivo evaluation and intestinal transport studies
Guihua Fang,
Bo Tang,
Yanhui Chao,
Yu Zhang*,
Hui Xu and
Xing Tang*
School of Pharmacy, Shenyang Pharmaceutical University, 103 Wenhua Road, Shenyang 110016, China. E-mail: tanglab@126.com; zywinner01@163.com; Fax: +86 24 23911736; Tel: +86 24 23986343
First published on 2nd November 2015
Abstract
The objective of the current study was to explore the potential of nanostructured lipid carriers (NLC) for oral delivery of docetaxel (DTX) and investigate the absorption mechanism in vivo. Docetaxel-loaded nanostructured lipid carriers (DNLCs) were prepared by emulsification–ultrasonication and their physicochemical properties were characterized. Differential scanning calorimetry (DSC) demonstrated that the drug was present in a amorphous state and FTIR analysis suggested that the interaction of DTX and lipids involved hydrogen bonding and hydrophobic interactions. DNLCs were found to be stable in simulated gastrointestinal fluids and in vitro drug release studies revealed that the formulation exhibited sustained drug release for 48 h by Fickian diffusion. The drug absorption in the intestine was markedly improved by NLCs. An in vivo pharmacokinetic study demonstrated that the AUC0–24 h for DNLCs (536.18 ± 91.21 ng mL−1 h) was increased 4.31-fold compared with that of docetaxel solution (DTX-Sol). DNLCs could be absorbed into the enterocytes through both endocytosis and passive transport. The oral bioavailability of DNLCs was significantly reduced after the lymphatic transport pathway was blocked. The overall results showed that the NLCs were a very effective method for increasing the oral absorption of docetaxel.
1. Introduction
Oral drug delivery has always been more popular than the parenteral route and has a number of advantages, including improvement in the quality of life of patients and reduced treatment costs.1,2 However, the majority of anticancer drugs have a low oral bioavailability, owing to their particular physicochemical properties, and physiological barriers, such as gastrointestinal instability, intestinal efflux and pre-systemic metabolism.3 Therefore, a means of increasing the bioavailability of anticancer drugs is a pre-requisite for the successful development of oral chemotherapy.Docetaxel (DTX) is a semisynthetic taxoid, structurally similar to paclitaxel, and is widely used for treating a broad range of human cancers, including refractory ovarian and breast cancer, non-small-cell lung carcinoma, head and neck carcinoma and leukemia.4–6 Currently, its commercial formulation, Taxotere®, is administered by the parenteral route. Oral administration of docetaxel results in extremely low bioavailability (less than 3%) owing to its acid degradation at low gastric pH, P-glycoprotein (P-gp) pump-mediated efflux in the apical membrane of intestinal epithelial cells, and cytochrome-P450 (CYP 450)-mediated metabolism in the liver or intestinal tract.7
In order to overcome the above-mentioned problems, nanotechnology-based drug delivery systems are considered as effective alternatives to improve the oral bioavailability of anticancer drugs.8 Nanocarriers have the ability to increase the drug solubility,9 prevent the drug from metabolic degradation within the gastrointestinal tract,10,11 and alter the drug absorption pathway.12 One of the most promising candidates for such a delivery system is nanostructured lipid carriers (NLCs).
NLCs consisting of solid lipids and liquid lipids are an advanced type of solid lipid nanoparticles, which offer the advantages of improving drug loading capacity and controlling release properties.13–15 Because the lipids are physiological and biodegradable, NLCs are also associated with an excellent biocompatibility compared with polymer-based nanocarriers. Furthermore, it is easier to scale up their production, and they are more stable during storage and less costly than liposomes. It has been reported that lipid-based formulations offer advantages after oral administration,16 such as lymphatic transport. Lymphatic delivery of drugs avoids the hepatic first-pass effect, and increases the plasma drug concentration, which is important for drugs that are highly metabolized in the liver. We are currently interested in increasing the oral bioavailability of DTX using NLCs and investigating their intestinal absorption mechanism involved.
In this work, docetaxel-loaded lipid carriers composed of Precifac ATO 5 (solid lipid) and MCT (liquid lipid) were prepared by an emulsification–ultrasonication technique. The prepared DNLCs were characterized for various physicochemical properties. The stability of DNLCs was evaluated in simulated gastric and intestinal fluid, and their in vitro release behavior was also investigated. The absorption of DNLCs in the stomach and intestine was investigated and compared with that of docetaxel solution. The in vivo oral pharmacokinetics and bioavailability of DNLCs were investigated in rats. In addition, the endocytosis pathways and their subsequent intestinal transport were explored to clarify the related mechanisms governing the oral absorption of DNLCs.
2. Methods
2.1. Materials and animals
Docetaxel and paclitaxel were obtained from Shanghai sanwei Pharma Ltd. Co., (Shanghai, China). Precifac ATO 5 (glyceryl palmitostearate) was kindly donated by Gattefosse (Lyon, France). Medium chain triglycerides (MCT), soybean lecithin (S 75) and egg lecithin (PC-98T) were obtained from Lipoid KG, (Ludwigshafen, Germany). Tween 80 was purchased from BASF (Ludwigshafen, Germany). Pepsin, sodium taurocholate and pancreatin were obtained from Sigma (St. Louis, MO, USA). Docetaxel solution was prepared based on the commercially available product, Taxotere® (10 mg mL−1 docetaxel in distilled water containing 25% (w/v) Tween 80 and 9.75% (v/v) ethanol).All of the animals studies were carried out according to the guidelines of the local Institutional Animal Ethical Care Committee (IAEC).
2.2. Preparation of DNLCs
DNLCs were prepared by emulsification–ultrasonication. In brief, Precifac ATO 5 (240 mg), Tween 80 (240 mg), MCT (60 mg), soybean lecithin (60 mg), and docetaxel (10 mg) were dissolved in 2 mL of ethanol at 75 °C in a water bath. The ethanol was removed by magnetic stirring for about 30 min and then, the aqueous solution (10 mL) was added dropwise to the melted lipid stirring for 5 min at 75 °C. The obtained primary emulsion was ultrasonicated using probe sonication at 400 W for 3 min with a 2 s pulse-on period and a 1 s pulse-off period. Finally, the nanoemulsion was cooled in an ice bath to allow the formation of DNLCs. The blank NLCs (BNLCs) were prepared using the same procedure, except for without docetaxel.2.3. Characterization of DNLCs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40, v/v). The flow rate was 1.0 mL min−1. The total amount of docetaxel incorporated into the DNLCs was determined by dissolving the formulation in ethanol and applying the HPLC method. The drug encapsulation efficiency of DNLCs was determined by ultrafiltration. The DNLCs were added into the ultrafiltration centrifuge tube (Millipore, MWCO: 100 kDa) and then centrifuged for 10 min at 3000 rpm. The centrifuged free docetaxel in the ultrafiltrate was determined by HPLC, as described above. The drug loading (DL, %) and entrapment efficiency (EE, %) of DNLCs were then calculated from eqn (1) and (2), respectively.
:40, v/v). The flow rate was 1.0 mL min−1. The total amount of docetaxel incorporated into the DNLCs was determined by dissolving the formulation in ethanol and applying the HPLC method. The drug encapsulation efficiency of DNLCs was determined by ultrafiltration. The DNLCs were added into the ultrafiltration centrifuge tube (Millipore, MWCO: 100 kDa) and then centrifuged for 10 min at 3000 rpm. The centrifuged free docetaxel in the ultrafiltrate was determined by HPLC, as described above. The drug loading (DL, %) and entrapment efficiency (EE, %) of DNLCs were then calculated from eqn (1) and (2), respectively.
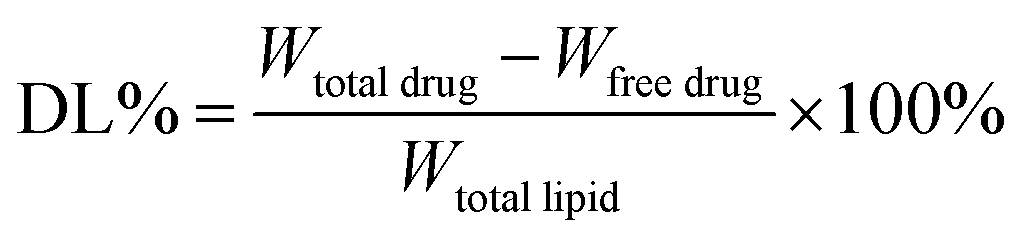 | (1) |
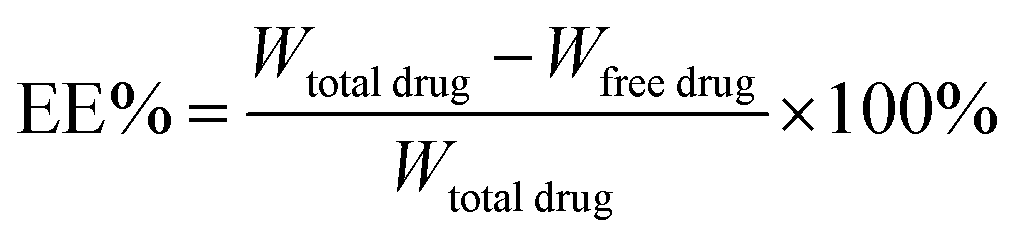 | (2) |
2.4. Differential scanning calorimetry (DSC)
The interaction of docetaxel with lipids and the physical state of docetaxel in the DNLCs were examined using a differential scanning calorimeter (Mettler-Toledo, Switzerland). Approximately 4 mg of the samples was weighed into an aluminum pan and sealed hermetically, and then the thermal analysis was conducted over the range of −10 to 200 °C at a heating rate of 10 °C min−1.2.5. Fourier transform infrared (FTIR) spectroscopy
FTIR spectra were obtained between 4000 and 400 cm−1 on a BRUKER IFS 55 FTIR system using the potassium bromide disk technique. The spectral resolution was 1 cm−1.2.6. Stability in simulated gastrointestinal (GI) fluids
The stability of DNLCs was evaluated in simulated gastric fluid (SGF) and simulated intestinal fluid (SIF). DTX solution was used as a control. The SGF consisted of 0.2% (w/v) NaCl, 0.32% (w/v) pepsin and HCl with a pH 1.2. To be consistent with the bioavailability assessment in the rats, a fasted-state buffer (pH 6.5) with lower enzyme concentration was selected as SIF. In brief, the SIF comprised 3 mM sodium taurocholate, 0.2 mM lecithin (PC-98T), 19.12 mM maleic acid, 34.8 mM NaOH, 68.62 mM NaCl and 1.7% (v/v) pancreatin. Pancreatin was freshly prepared by dissolving 1 g of pancreatin powder in 5 mL of fasted-state buffer, centrifuging at 16000 rpm for 15 min, and separating supernatant. To begin the study, 2.5 mL of DNLCs was added to 10 mL SGF or SIF. The mixture was incubated at 37 °C with a shaking speed of 100 rpm. The particle size, drug content and pH were measured at predetermined time points.
2.7. In vitro release
In vitro release of docetaxel from DNLCs was performed using a dialysis technique. In brief, 1 mL samples of the docetaxel ethanol solution (docetaxel dissolved in ethanol), docetaxel solution and DNLCs dispersion were sealed in pre-swelled dialysis bags (MWCO: 14 kDa). The bags were immersed in 20 mL release medium (phosphate buffer solution containing 0.5% Tween-80 w/v, pH 6.8), then placed in a water bath shaker at a rate of 100 rpm at 37 ± 0.5 °C. A total of 20 mL of sample was removed at designated intervals, followed by addition of the same volume of fresh release medium. The amount of docetaxel released was determined by HPLC as described above. To determine the docetaxel release mechanism, the in vitro release data obtained from DNLCs were fitted to zero-order, first-order, Higuchi and Ritger–Peppas models. Model equations are as follow.Zero-order model: Q = k0t.
First-order model: ln(100 − Q) = −k1t.
Higuchi models: Q = k2t1/2.
Ritger–Peppas model: Q = ktn.Where Q is the fraction of drug release at time (t), k0, k1, k2 and k is a rate constant, n is the release exponent.
2.8. Absorption of DNLCs in the GI tract
The absorption of DNLCs in the GI tract was investigated in rats by an in situ method.18 Male Sprague-Dawley (SD) rats weighing 180–220 g were fasted 18 h before the experiments. Anesthesia was induced by intraperitoneal injection of 20% urethane (1 g kg−1), and the rats were kept in a supine position at 37 °C. A small middle incision was carefully made in the abdomen and, in order to evaluate the DNLCs absorption in the stomach, the stomach was exposed and the pylorus was ligated, then the DNLCs were intragastrically administered to the rats at 15 mg kg−1. Similarly, in order to evaluate the DNLCs absorption in the intestine, the DNLCs were given to the rats by intraduodenal administration at the same dose. Docetaxel solution was used for the control groups. After administration, the incision was closed by stitching, and the rats were kept under infrared lamps to maintain a constant body temperature. Blood samples were taken from the retrobulbar venous plexus into heparinized tubes at 0.25, 0.5, 0.75, 1, 2 h and, then, plasma was separated immediately by centrifugation at 8000 rpm for 10 minutes and stored at −20 °C for analysis.The UPLC-MS/MS method was applied for the determination of docetaxel. The plasma samples were processed by liquid–liquid extraction.17 Briefly, 100 μL plasma was mixed with 20 μL of the paclitaxel solution (internal standard) and vortexed for 1 min. Then, 2.5 mL of tert butyl methyl ether was added to the mixture, vortexed for another 10 min, followed by centrifugation at 4000 rpm for 10 min. Then, 2 mL of the supernatant was transferred to a clean tube and evaporated to dryness. One hundred microliters of methanol was added to the residue, vortexed for 10 min and centrifuged at 12000 rpm for another 10 min, then 5 μL of supernatant was injected into the UPLC-MS/MS system (Waters Corp., Milford, MA, USA) for measurement. Analytical data were acquired using MassLynx software.
The chromatographic separation was carried out on a C18 column (ACQUITY UPLC™ BEH C18, 50 mm × 2.1 mm, 1.7 μm, Waters Corp, Milford, MA, USA) at 40 °C. The mobile phase consisted of A (acetonitrile) and B (deionized water containing 5 mM ammonium acetate). Mass spectrometric detection was performed in positive ESI mode. The capillary voltage was 2.8 kV, the collision energy was 8 V, and the ion source temperature was 150 °C. Quantification was carried out in multiple reaction monitoring (MRM) for docetaxel [M + H]+ m/z 808.68 and paclitaxel [M + H]+ m/z 854.64, respectively. Samples were quantitatively determined using a calibration curve over the range 5–500 ng mL−1. The correlation coefficient was 0.990.
2.9. In vivo pharmacokinetic behavior in rats
The male SD rats were fasted 18 h before the experiments with free access to water and randomly divided into two groups, then given docetaxel solution and DNLCs at 15 mg kg−1 perorally. Blood samples were collected from the retrobulbar venous plexus of rats into heparinized tubes at 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 10, 12 and 24 h after administration. Plasma was separated by centrifugation (8000 rpm, 10 min) and analyzed by UPLC-MS/MS as described above.2.10. Mechanism investigation
2.11. In vivo biocompatibility study
To evaluate the intestinal toxicity caused by NLCs, an histopathologic examination was performed.20 Blank NLCs were given orally to rats at dose of 150 mg kg−1, 450 mg kg−1, 900 mg kg−1. Saline was used as a control. After 2 h and 6 h, the rats were sacrificed. The intestine was removed and fixed in 10% formalin, stained with hematoxylin – eosin. The prepared samples were observed under a light microscope.2.12. Statistical analysis
All the data were expressed as mean values ± SD. Student's t-test was used to investigate statistical differences. Statistical difference was defined as p < 0.05, and a significant difference was set at p < 0.01.3. Results
3.1. Preparation and characterization of DNLCs
The DNLCs had a small particle size (85.8 ± 4.7 nm), and a narrow size distribution (0.242 ± 0.045) (Fig. 1A). The zeta potential of the DNLCs and BNLCs was −8.88 ± 0.37, −9.87 ± 1.07 mV, respectively, and the negative zeta potential might be due to the soybean lecithin being negatively charged in aqueous solution. The entrapment efficiency and drug loading of DNLCs was 99.89 ± 0.01% and 3.07 ± 0.01%, respectively. A TEM image of the DNLCs shows spherical particles with a smooth surface (Fig. 1B).
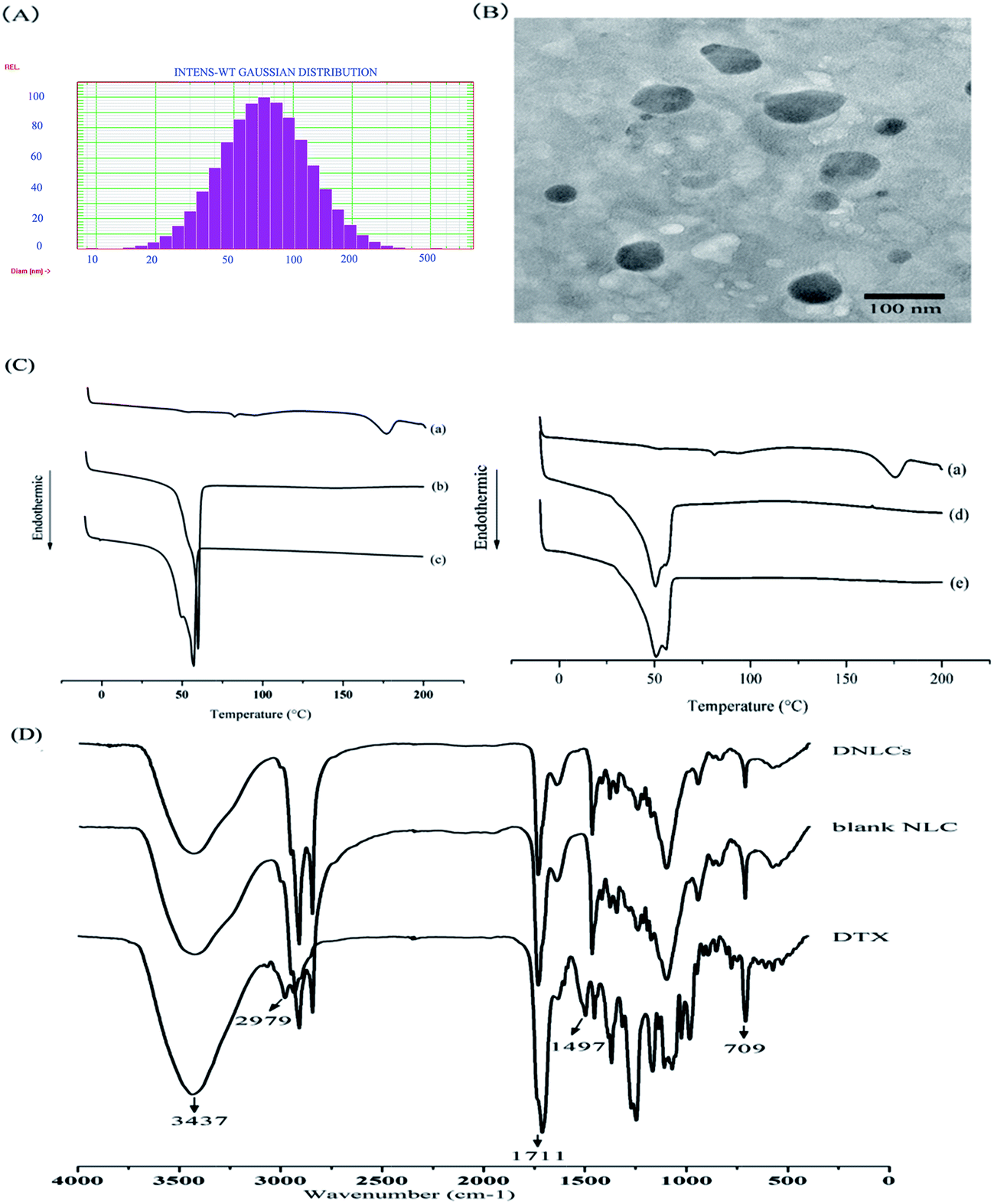 | ||
| Fig. 1 Size distribution (A) and TEM image (B) of DNLCs. DSC thermograms (C) of DTX (a), Precifac ATO 5 (b), the mixture of DTX, Precifac ATO 5 and MCT (c), blank NLC (d) and DNLCs (e). FTIR spectra (D) of DNLCs, blank NLCs and DTX. | ||
3.2. Differential scanning calorimetry
The DSC thermograms of docetaxel, Precirol ATO 5 and the mixture of docetaxel with Precirol ATO 5 and MCT are shown in Fig. 1C, docetaxel showed a characteristic crystalline form melting peak at 175.8 °C and the docetaxel endothermic peak had disappeared in the mixture of docetaxel and lipid excipients, suggesting that there was an interaction between docetaxel and the lipid excipients. Comparing the endothermic peak between Precirol alone and the mixture of docetaxel and lipid excipients, the DSC thermogram of Precirol ATO 5 showed a sharp endothermic peak at 59.8 °C, and there was a depression in the endothermic peak following the addition of MCT and docetaxel. The depression could be correlated with the dissolution of MCT and docetaxel in Precirol ATO 5 with the formation of less ordered structure.21 In addition, it was observed that the docetaxel endothermic peak was absent in the DNLCs (Fig. 1C), indicating that the drug molecules inside the DNLCs were in an amorphous or molecular state.3.3. Fourier transform infrared spectroscopy (FTIR) analysis
FTIR was used to investigate the intermolecular interaction between DTX and lipid excipients. The position of the absorption bands of DTX at 3437 (O–H and N–H stretching vibrations) and 1711 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations) cm−1 were absent in the DNLCs (Fig. 1D), indicating that hydrogen bonds are formed between docetaxel and lipid excipients, which containing carbonyl groups (CO, hydrogen receptor) and hydroxyl groups (O–H, hydrogen donor). The absence of absorption bands at 2979 (C–H stretching vibrations), 1497 (aromatic CC stretching vibration) and 709 (aromatic C–H out-of-plane bending) cm−1 of docetaxel indicated that a hydrophobic interaction could be another intermolecular force between docetaxel and aliphatic chains of lipid excipients.
O stretching vibrations) cm−1 were absent in the DNLCs (Fig. 1D), indicating that hydrogen bonds are formed between docetaxel and lipid excipients, which containing carbonyl groups (CO, hydrogen receptor) and hydroxyl groups (O–H, hydrogen donor). The absence of absorption bands at 2979 (C–H stretching vibrations), 1497 (aromatic CC stretching vibration) and 709 (aromatic C–H out-of-plane bending) cm−1 of docetaxel indicated that a hydrophobic interaction could be another intermolecular force between docetaxel and aliphatic chains of lipid excipients.
3.4. Stability in simulated gastrointestinal fluids
It has been reported that the gastrointestinal fluid volume of the rats was low (<10 mL) and the secretion rate of bile was slow (∼1.5 mL h−1).22 Therefore, in this case, the volume of the simulated gastrointestinal fluids was set at 10 mL. As shown in Fig. 2, the particle size and pH were almost unchanged following incubation in SGF for 2 h. However, the drug content decreased about 11%. This may be related to the presence of few drugs on the surface of the nanoparticles, which would diffuse across the hydrophilic shell and be degraded by acid. Since digestion of the lipid mainly occurred in the intestine due to the presence of the pancreatin, the stability of DNLCs in the SIF was also studied. The particle size, drug content and pH were almost unchanged within 6 h incubation. It has been reported that lipids could be digested by pancreatin, producing fatty acids and mono/diglycerides.23 Fatty acids liberation therefore resulted in a drop in pH. In our case, the pH value of SIF was retained at ∼6.7, which indicated that the outer Tween 80 with PEG could protect lipid core from enzymatic degradation. In addition, NLCs could improve the stability of docetaxel in SGF due to the encapsulation.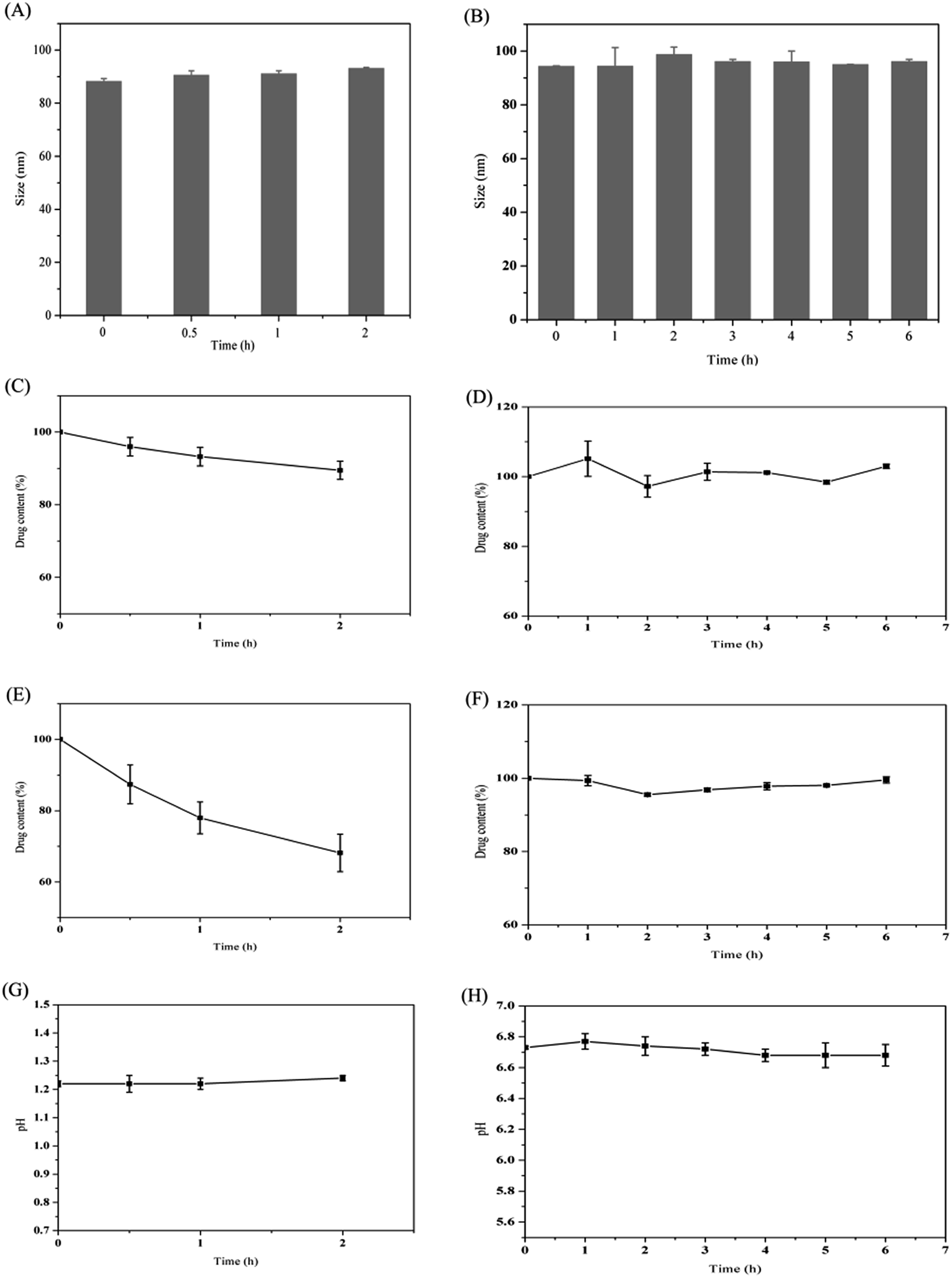 | ||
| Fig. 2 DNLCs particle size following incubation in SGF (A) and SIF (B). Drug content of DNLCs following incubation in SGF (C) and SIF (D). Drug content of DTX solution following incubation in SGF (E) and SIF (F), the initial drug content was set as 100%. pH value of SGF (G) and SIF (H) following incubation with DNLCs. | ||
3.5. In vitro release
The in vitro drug release profiles of different formulations are shown in Fig. 3. In the case of the docetaxel ethanol solution, 100% docetaxel had been released at the end of 8 h indicating that the cellulose membrane was not a limiting factor in the controlled release of docetaxel from the DNLCs formulation. The docetaxel solution exhibited relatively rapid drug release with 51% in 2 h and 62% in 8 h. In contrast, DNLCs exhibited sustained drug release with 60% after 8 h and 78% after 48 h. The correlation coefficient value of different release models are calculated and summarized in Table 1. It was found that DNLCs preferentially followed the Ritger–Peppas model. The release exponent n was 0.363 (n < 0.45), which suggested Fickian diffusion as the mechanism of drug release.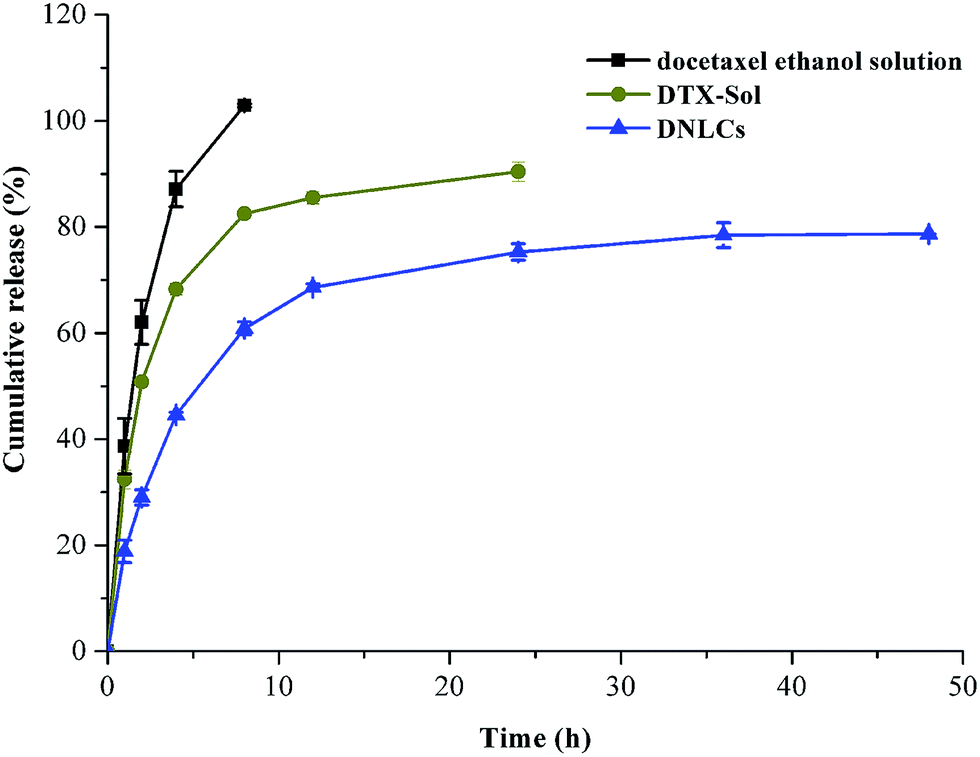 | ||
| Fig. 3 In vitro release profile of DTX from DTX ethanol solution, DTX-Solution, DNLCs in phosphate buffered solution (0.5% of Tween 80 in PBS, pH 6.8) at 37 ± 0.5 °C (n = 3). | ||
3.6. Absorption of DNLCs in the GI tract
The absorption of DNLCs in the GI tract was investigated in rats using an in situ method, with docetaxel solution as control group and the AUC0–2 h was calculated. After intragastric administration, the docetaxel concentration in plasma was undetectable in the case of both DNLCs and docetaxel solution, which suggested that the stomach was not a good absorption segment for docetaxel, and nanostructured lipid carriers could not improve docetaxel absorption in the stomach. In addition, the docetaxel plasma concentration was determined at 15, 30, 45, 60, 90, 120 min after intraduodenal administration, and the results obtained indicated that the docetaxel concentration from DNLCs was higher at each time point compared with docetaxel solution (Fig. 4). The AUC0–2 h of DNLCs and docetaxel solution was 89.56 ± 15.99 and 20.59 ± 12.74 ng mL−1 h, respectively. Compared with docetaxel solution, the AUC0–2 h of DNLCs was significantly increased 4.35-fold. These results indicated that the absorption of docetaxel from the intestinal tract could be markedly increased after encapsulation into nanostructured lipid carriers.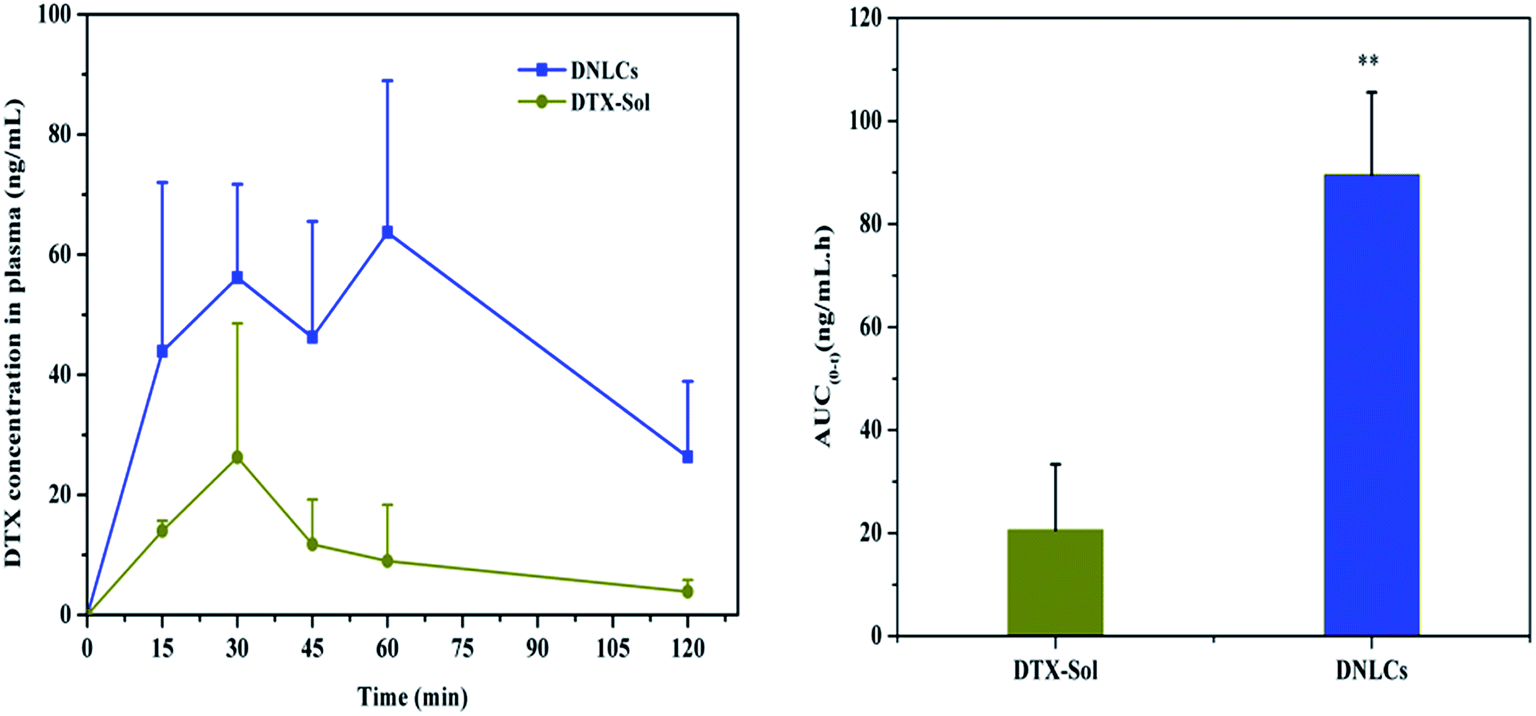 | ||
| Fig. 4 The absorption of DNLCs in the intestinal tract compared with DTX solution (15 mg kg−1). The plasma concentration (A) and the AUC0–2 h of docetaxel in rats after intraduodenal administration (B) (n = 3). **, p < 0.01. | ||
3.7. In vivo pharmacokinetics in rats
In order to further investigate the enhancement of docetaxel oral bioavailability by nanostructured lipid carriers, we performed pharmacokinetic studies using docetaxel solution and DNLCs. The plasma concentration of docetaxel versus time profile is illustrated in Fig. 5. The main pharmacokinetic parameters were calculated by DAS 2.0 software.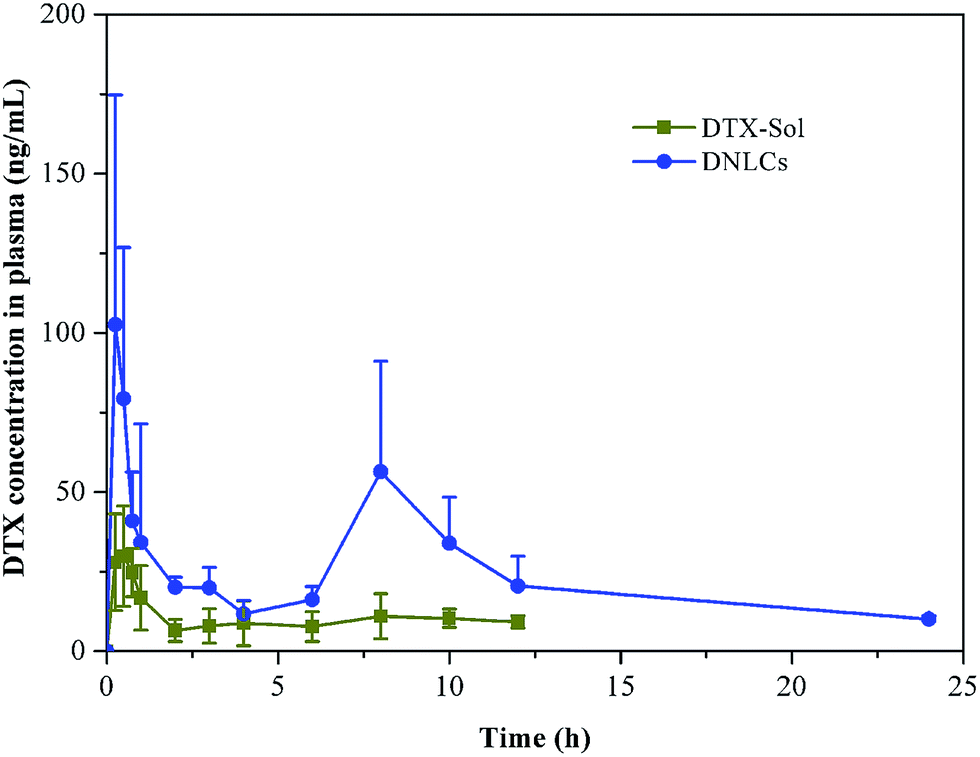 | ||
| Fig. 5 Mean plasma concentration–time curves of DTX in rats after oral administration of different dosage forms of DTX. Indicated values are the means (SD) of six parallel animals. | ||
The results showed that there was a significant difference in the main pharmacokinetic parameters of AUC0–24 h and MRT between docetaxel solution and DNLCs. The AUC0–24 h of DNLCs (536.18 ± 91.21 ng mL−1 h) was markedly improved 4.31-fold in comparison with docetaxel solution (124.35 ± 27.87 ng mL−1 h), the MRT of DNLCs (9.71 ± 0.91 h) was also increased 1.72-fold compared with that of docetaxel solution (5.64 ± 0.46 h), likely due to the sustained release of docetaxel from DNLCs.
3.8. Uptake of DNLCs into rat everted gut sacs
It has been reported that the everted gut sac model could be applied to evaluate the absorption of many drugs in the intestine.24,25 Because the intestinal tissue are from the rats, the experimental data obtained can be correlated well with in vivo experiments. Previous reports have shown that nanostructured lipid carriers can be transported across the intestinal epithelial cells by both clathrin- and caveolae-mediated endocytosis.26 Hence, the uptake of DNLCs in the intestine was investigated. Firstly, the absorption of DNLCs in the everted intestinal rings was performed at 4 and 37 °C. It has been reported that lowering the temperature could inhibit the active uptake like endocytosis.27 As shown in Fig. 6, at 4 °C, the uptake of DNLCs was only about 13% of that at 37 °C. This result indicated that endocytosis was predominant for the absorption of DNLCs in the intestine. To further clarify the uptake pathway of DNLCs in enterocytes, the uptake experiments were carried out in the presence of specific inhibitors for different types of endocytosis. The results are shown in Fig. 6, and it can be seen that different types of endocytosis inhibitors had different effects on the intestinal absorption. Incubation with DNLCs in the presence of chlorpromazine, a clathrin-mediated endocytosis inhibitor that inhibits a RhoGTPase for the formation of clathrin-coated vesicles,28 indomethacin, a caveolae-mediated endocytosis inhibitor by accumulating arachidonate,29 and colchicine, a macropinocytosis inhibitor that depolymerizes microtubules,30 resulted in a significant decrease of in the intestinal absorption of DNLCs, which indicated that clathrin, caveolae and macropinocytosis are involved in the endocytosis pathway. In contrast, quercetin, a caveolae- and clathrin-independent endocytosis inhibitor,31 had no effect on the absorption of DNLCs, implying that the endocytosis of DNLCs was not via caveolae- and clathrin-independent endocytosis.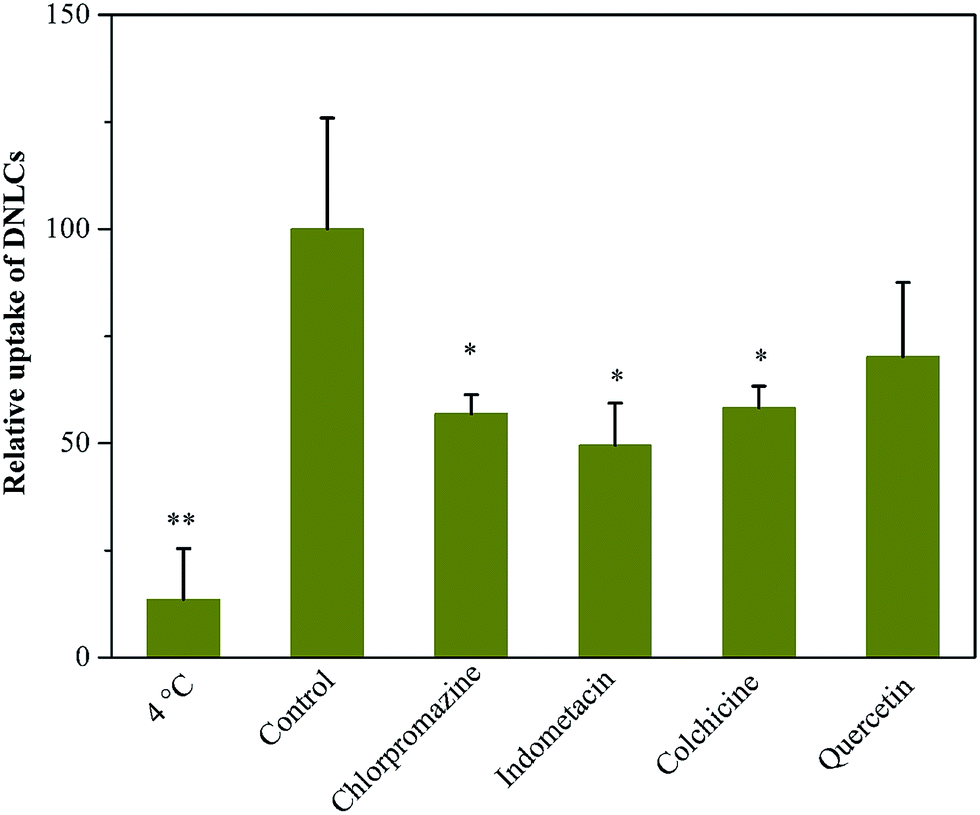 | ||
| Fig. 6 The endocytosis pathways of DNLCs after incubation with endocytosis inhibitors at 37 °C (control) or 4 °C (n = 3), *, p < 0.05; **, p < 0.01. | ||
3.9. Intestinal lymphatic transport of DNLCs in rats
Following DNLCs absorption into intestinal enterocytes, they entered the systemic circulation via the portal vein or intestinal lymphatic. In order to elucidate the intestinal transport pathway, rats were treated with the lymphatic transport inhibitor, cycloheximide. It has been reported that cycloheximide can inhibit the lymphatic transport pathway without damaging other passive and active absorption pathways.32 When the rats were treated with cycloheximide, the peak concentration of DTX was dramatically decreased and the AUC0–12 h was reduced by about 57% (p < 0.05) in comparison with the control group (Fig. 7). This result demonstrated the importance of intestinal lymphatic transport in the oral absorption of DNLCs.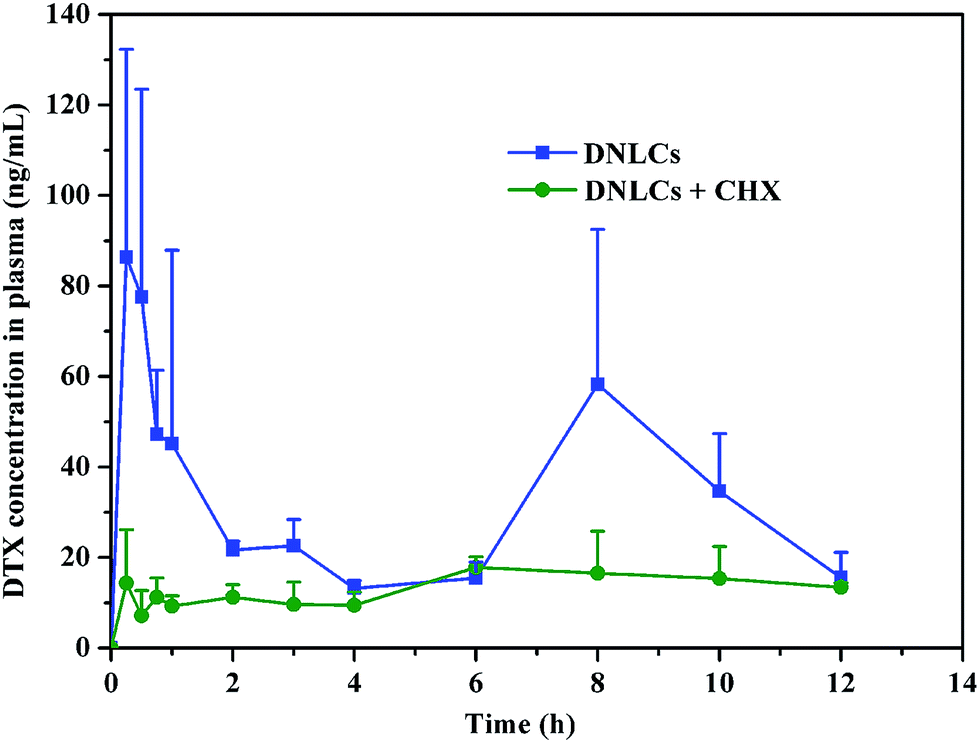 | ||
| Fig. 7 The plasma docetaxel concentration–time profiles (±SD) obtained following oral administration of 15 mg kg−1 DNLCs to non-treated rats, and to cycloheximide-treated rats 1 h before administration (n = 4). | ||
3.10. In vivo biocompatibility study
Histological study showed that no significant changes in the structures of the intestine exposed to different concentration of blank NLCs in comparison with control (Fig. 8). The mucosal erosions and disruption of the intestinal epithelium cells were not observed in all samples, which indicated that the amounts of blank NLCs used did not cause any harm to the intestine. Therefore, the NLCs demonstrated excellent biocompatibility in vivo and could be considered as safe for oral drug administration purposes.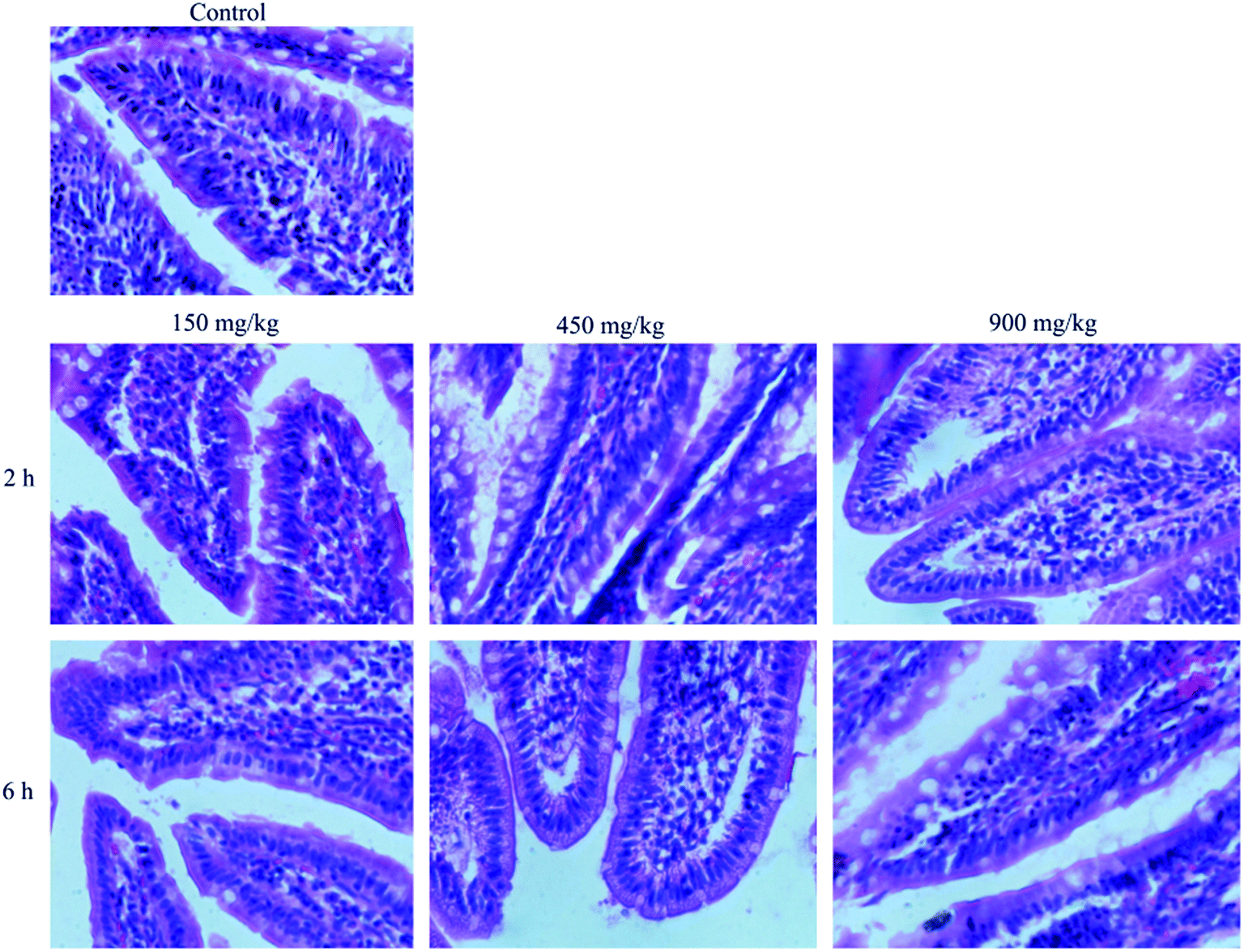 | ||
| Fig. 8 Morphology of intestinal mucosa of rats after oral administration of physiological saline (control), different concentration of BNLCs at 2 h and 6 h. | ||
4. Discussion
In the recent years, lipid-based drug delivery systems have demonstrated obvious advantages in improving the oral bioavailability of BCS II drugs.19,33,34 Liu et al. designed nanostructured lipid carriers for intravenous delivery of docetaxel to improve the therapeutic efficacy in tumor.35 In comparison with parental drug delivery, the oral administration is considered to be a preferred route. In general, oral delivery has been shown to exhibit several advantages over the intravenous injection. On the one hand, the oral administration can be conducted at home, which is convenient and can reduce the costs of administration. On the other hand, it avoids the discomfort of injection, improving patient compliance. In the present work, we found that NLCs were potential vehicles for the oral delivery of water-insoluble anticancer drugs such as docetaxel and explored their possible absorption mechanism in vivo.DNLCs were produced by emulsification–ultrasonication, taking into account the chemical structure, and the special affinity between docetaxel and lipid excipients that might be present. The DSC results indicated that there was a strong interaction between them. The FTIR results further supported that the drug association with lipids was due to intermolecular hydrogen bonds and hydrophobic interactions. The liquid state of MCT oil allows lipid carriers to encapsulate higher amounts of docetaxel and reduces the particle crystallinity which imparts better stability. The total docetaxel content assay results indicated that approximately 1.0 mg mL−1 of docetaxel was present in the NLCs, which was far higher than its water solubility of 4.95 μg mL−1.36 Moreover, the average size of DNLCs was 85.8 nm. It has been reported that particles with a size below 200 nm could effectively diffuse through the mucus and avoid elimination by mucociliary clearance.37 In addition, the results obtained indicated that the DNLCs were very stable in the SGF and SIF. Accordingly, the greatly increased aqueous solubility of docetaxel by DNLCs and their good stability in GI fluids and small particle size will facilitate their transport across the intestinal barrier.
In pharmacokinetic study, it was interesting to find that two peaks of DNLCs were observed (Fig. 5). The first peak of DNLCs in blood took place during 0–30 min, which indicated that nanoparticles were absorbed quickly from the intestinal tract into blood. The docetaxel concentration then began to decrease due to the elimination of nanoparticles, which might be related to the uptake by the phagocytic cells of the reticuloendothelial system (RES).38 Docetaxel would transport across the phagocytic cell membrane and diffuse into blood due to its high lipophilicity.39 As a result, the plasma drug concentration was increased, which was related to the second peak occurring at about 6–10 h. The similar phenomenon was also observed by Yuan et al.'s group.40 Overall, the in vivo pharmacokinetic behavior indicated that the oral bioavailability of docetaxel was significantly improved after incorporation into NLCs.
For a better understanding of the intestinal absorption of DNLCs, several endocytosis inhibitors were used to explore the transport mechanisms. The data obtained from this experiment suggested that endocytosis attached much importance in the absorption of DNLCs. In addition, the results of the in vitro release study suggested that docetaxel could gradually diffuse from the DNLCs in the intestine under sink conditions. Based on a comprehensive analysis, both passive diffusion of free soluble docetaxel and endocytosis of DNLCs could be involved in the absorption of DNLCs in the intestine.
The results of the intestinal lymphatic drug transport indicate that the lymphatic pathway contributes to the transport of docetaxel into the systemic circulation. On the basis of the results of the intestinal uptake of DNLCs, the NLCs was internalized into intestinal epithelium cells through endocytosis (clathrin-mediated, caveolae-mediated and macropinocytosis). The transport pathways of lipid nanoparticles across the simulated intestinal epithelial monolayer has been reported in the literature. Chai et al.41 reported that the lipid nanoparticles were internalized into intestinal epithelium cells, some nanoparticles could be transported to ER (endoplasmic reticulum) and Golgi complex through AEE (apical recycling endosomes)/CRE (common recycling endosome) route, followed by stimulating chylomicrons formation and exocytosis. Because chylomicrons usually have larger size, they preferentially access highly permeable lymphatics and then transported into blood.42 Intestinal lymphatic drug transport exhibits many advantages over oral absorption via the portal vein. In particular, this is very advantageous for most anticancer drugs such as docetaxel that are highly metabolized in the liver. Drugs are transported into the systemic circulation via the intestinal lymphatic system, which could bypass the first-pass effect. Therefore, the oral bioavailability of docetaxel could be increased.
On basis of the present research, a hypothetical absorption mechanism for DNLCs in the intestinal tract is present in Fig. 9. DNLCs exhibit gastrointestinal transit to the intestine, and some drugs release from the DNLCs, and passively diffuse across the enterocytes to enter the portal vein, while other intact DNLCs are internalized into the enterocytes, then transported to the systemic circulation via the portal vein and lymphatic pathway. Three mechanisms contribute to the enhanced oral absorption of docetaxel by NLCs. Firstly, the in vitro characteristics confirm the small size (<100 nm) and good stability of DNLCs in simulated GI fluids, which could be beneficial for the diffusion across the barrier of unstirred water layer to absorptive enterocytes. Secondly, DNLCs could be directly internalized to the intestinal epithelium cell bypassing the P-gp efflux pumps. In addition, DNLCs could be transported to the systemic circulation via the intestinal lymphatic pathway, thus avoiding first-pass metabolism.
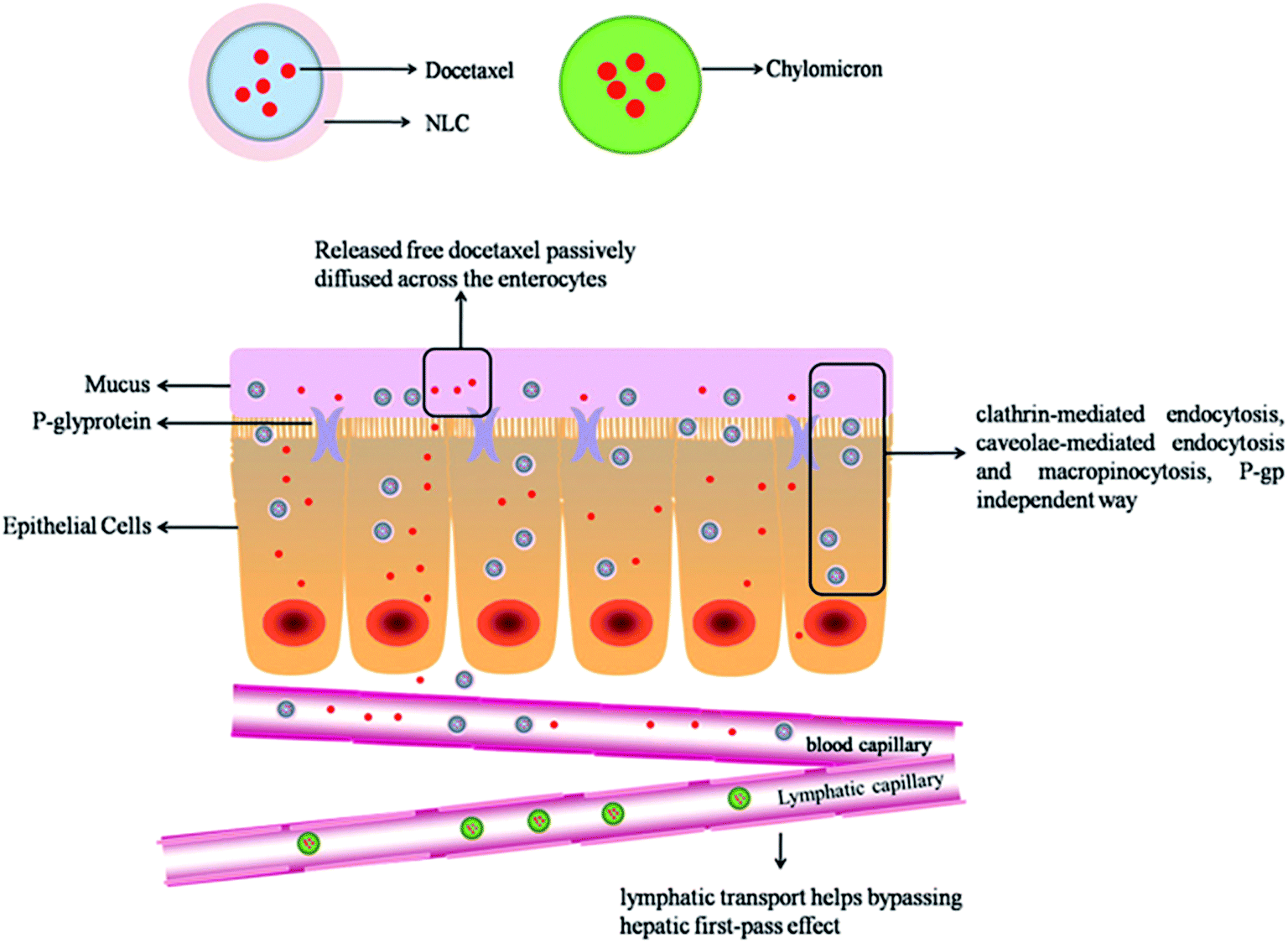 | ||
| Fig. 9 Illustration of the hypothetic absorption process for DNLCs. | ||
5. Conclusion
In summary, nanostructured lipid carriers were designed to improve the oral absorption of docetaxel. The DNLCs achieved excellent drug entrapment, a satisfactory particle size and good GI stability. The absorption of DNLCs in the intestinal segment was markedly improved compared with docetaxel solution. After oral administration of DNLCs, the relative bioavailability was improved 4.31-fold compared with that of docetaxel solution. Both endocytosis and passive transport could be involved in the absorption of DNLCs. Lymphatic transport contributes to improving the absorption of docetaxel. In vivo biocompatibility study showed that NLCs had a low toxicity with mucosal damage. The overall results indicate that the NLCs are very promising method for enhancing the oral absorption of anticancer drugs such as docetaxel.Acknowledgements
This work is supported by the National High-tech R&D Program of China 863 Program (2012AA020305) and the National Basic Research Program of China 973 Program (2009CB930300). Dr David B. Jack is gratefully thanked for correcting the manuscript.References
- V. J. O'Neill and C. J. Twelves, Br. J. Cancer, 2002, 87, 933 CrossRef PubMed.
- A. H. Partridge, J. Avorn, P. S. Wang and E. P. Winer, J. Natl. Cancer Inst., 2002, 94, 652 CrossRef PubMed.
- K. Thanki, R. P. Gangwal, A. T. Sangamwar and S. Jain, J. Controlled Release, 2013, 170, 15 CrossRef CAS PubMed.
- D. L. Ornstein and J. R. Rigas, Oncologist, 1998, 3, 86 CAS.
- I. C. Smith, S. D. Heys, A. W. Hutcheon, I. D. Miller, S. Payne, F. J. Gilbert, A. K. Ah-See, O. Eremin, L. G. Walker, T. K. Sarkar, S. P. Eggleton and K. N. Ogston, J. Clin. Oncol., 2002, 20, 1456 CrossRef CAS PubMed.
- A. I. Dreyfuss, J. R. Clark, C. M. Norris, R. M. Rossi, J. W. Lucarini, P. M. Busse, M. D. Poulin, L. Thornhill, R. Costello and M. R. Posner, J. Clin. Oncol., 1996, 14, 1672 CAS.
- K. Hu, S. Cao, F. Hu and J. Feng, Int. J. Nanomed., 2012, 7, 3537 CrossRef CAS PubMed.
- C. Luo, J. Sun, Y. Du and Z. He, J. Controlled Release, 2014, 176, 94 CrossRef CAS PubMed.
- V. Zabaleta, G. Ponchel, H. Salman, M. Agueros, C. Vauthier and J. M. Irache, Eur. J. Pharm. Biopharm., 2012, 81, 514 CrossRef CAS PubMed.
- S. Jain, D. Kumar, N. K. Swarnakar and K. Thanki, Biomaterials, 2012, 33, 6758 CrossRef CAS PubMed.
- S. Jain, S. R. Patil, N. K. Swarnakar and A. K. Agrawal, Mol. Pharm., 2012, 9, 2626 CrossRef CAS PubMed.
- D. Zhang, X. Pan, S. Wang, Y. Zhai, J. Guan, Q. Fu, X. Hao, W. Qi, Y. Wang, H. Lian, X. Liu, Y. Sun, Z. He and J. Sun, Mol. Pharm., 2015, 12, 2337 CrossRef CAS PubMed.
- M. L. Bondi, E. F. Craparo, G. Giammona, M. Cervello, A. Azzolina, P. Diana, A. Martorana and G. Cirrincione, Drug Delivery, 2007, 14, 61 CrossRef CAS PubMed.
- S. Doktorovova, J. Araujo, M. L. Garcia, E. Rakovsky and E. B. Souto, Colloids Surf., B, 2010, 75, 538 CrossRef CAS PubMed.
- F. Han, S. Li, R. Yin, H. Liu and L. Xu, Colloids Surf., A, 2008, 315, 210 CrossRef CAS.
- H. Harde, M. Das and S. Jain, Expert Opin. Drug Delivery, 2011, 8, 1407 CrossRef CAS PubMed.
- G. Fang, B. Tang, Z. Liu, J. Gou, Y. Zhang, H. Xu and X. Tang, Eur. J. Pharm. Sci., 2014, 60, 1 CrossRef CAS PubMed.
- H. Li, X. Zhao, Y. Ma, G. Zhai, L. Li and H. Lou, J. Controlled Release, 2009, 133, 238 CrossRef CAS PubMed.
- Z. Zhang, H. Bu, Z. Gao, Y. Huang, F. Gao and Y. Li, Int. J. Pharm., 2010, 394, 147 CrossRef CAS PubMed.
- Y. Zhang, X. Wu, L. Meng, R. Ai, N. Qi, H. He, H. Xu and X. Tang, Int. J. Pharm., 2012, 436, 341 CrossRef CAS PubMed.
- F. Li, Y. Wang, Z. Liu, X. Lin, H. He and X. Tang, Drug Dev. Ind. Pharm., 2010, 36, 508 CrossRef CAS PubMed.
- M. U. Anby, T. H. Nguyen, Y. Y. Yeap, O. M. Feeney, H. D. Williams, H. Benameur, C. W. Pouton and C. J. Porter, Mol. Pharm., 2014, 11, 4069 CrossRef CAS PubMed.
- Y. Ting, Y. Jiang, Y. Lan, C. Xia, Z. Lin, M. A. Rogers and Q. Huang, Mol. Pharm., 2015, 12, 2229 CrossRef CAS PubMed.
- Q. Fu, J. Sun, X. Ai, P. Zhang, M. Li, Y. Wang, X. Liu, Y. Sun, X. Sui, L. Sun, X. Han, M. Zhu, Y. Zhang, S. Wang and Z. He, Int. J. Pharm., 2013, 448, 290 CrossRef CAS PubMed.
- H. Yuan, C. Y. Chen, G. H. Chai, Y. Z. Du and F. Q. Hu, Mol. Pharm., 2013, 10, 1865 CrossRef CAS PubMed.
- A. Beloqui, M. A. Solinis, A. R. Gascon, A. del Pozo-Rodriguez, A. des Rieux and V. Preat, J. Controlled Release, 2013, 166, 115 CrossRef CAS PubMed.
- J. Casley-Smith, J. Microsc., 1969, 90, 15 CrossRef.
- R. Firdessa, T. A. Oelschlaeger and H. Moll, Eur. J. Cell Biol., 2014, 93, 323 CrossRef CAS PubMed.
- M. Gumbleton, A.-n. G. Abulrob and L. Campbell, Pharm. Res., 2000, 17, 1035 CrossRef CAS.
- J. Linares, M. C. Matesanz, M. Vila, M. J. Feito, G. Goncalves, M. Vallet-Regi, P. A. Marques and M. T. Portoles, ACS Appl. Mater. Interfaces, 2014, 6, 13697 CAS.
- J. Wang, L. Li, Y. Du, J. Sun, X. Han, C. Luo, X. Ai, Q. Zhang, Y. Wang, Q. Fu, Z. Yang and Z. He, Mol. Pharm., 2015, 12, 463 CrossRef CAS PubMed.
- A. Dahan and A. Hoffman, Eur. J. Pharm. Sci., 2005, 24, 381 CrossRef CAS PubMed.
- F. Gao, Z. Zhang, H. Bu, Y. Huang, Z. Gao, J. Shen, C. Zhao and Y. Li, J. Controlled Release, 2011, 149, 168 CrossRef CAS PubMed.
- Z. Zhang, J. Huang, S. Jiang, Z. Liu, W. Gu, H. Yu and Y. Li, J. Pharm. Sci., 2013, 102, 1301 CrossRef CAS PubMed.
- D. Liu, Z. Liu, L. Wang, C. Zhang and N. Zhang, Colloids Surf., B, 2011, 85, 262 CrossRef CAS PubMed.
- K. Gao, J. Sun, K. Liu, X. Liu and Z. He, Drug Dev. Ind. Pharm., 2008, 34, 1227 CrossRef CAS PubMed.
- L. Plapied, N. Duhem, A. des Rieux and V. Préat, Curr. Opin. Colloid Interface Sci., 2011, 16, 228 CrossRef CAS.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505 CrossRef CAS PubMed.
- Y. Wang, X. Li, L. Wang, Y. Xu, X. Cheng and P. Wei, Int. J. Nanomed., 2011, 6, 1497 CAS.
- H. Yuan, J. Chen, Y. Z. Du, F. Q. Hu, S. Zeng and H. L. Zhao, Colloids Surf., B, 2007, 58, 157 CrossRef CAS PubMed.
- G. H. Chai, F. Q. Hu, J. Sun, Y. Z. Du, J. You and H. Yuan, Mol. Pharm., 2014, 11, 3716 CrossRef CAS PubMed.
- J. A. Yanez, S. W. Wang, I. W. Knemeyer, M. A. Wirth and K. B. Alton, Adv. Drug Delivery Rev., 2011, 63, 923 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |