
The nature of interfacial binding of imidazole and carbene ligands with M20 nanoclusters (M = Au, Ag and Cu) – a theoretical study†
K. R. Geethalakshmi*ab,
Xia Yangc,
Qiao Sund,
T. Y. Ngb and
D. Wangc
aCentro de Física de Materiales (CSIC-UPV/EHU) and Donostia International Physics Center (DIPC), P. Manuel de Lardizabal 5, 20018 Donostia, Spain. E-mail: krgeetha@ntu.edu.sg
bSchool of Mechanical and Aerospace Engineering, Nanyang Technological University Singapore, 50 Nanyang Avenue, Singapore-639798
cInstitute of High Energy Physics, Chinese Academy of Science, Beijing, 100049, China
dInstitute of Quantitative Biology and Medicine, Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, School of Radiation Medicine and Protection, Medical College of Soochow University, Soochow University, Suzhou 215123, China
First published on 12th October 2015
Abstract
To inspire more exciting developments in the design and advances of self-assembled monolayers (SAMs), the fundamental understanding of the nature of interaction between metal nanoparticles and certain functional groups is very crucial. In this work, the interactions of imidazole based organic ligands with metal clusters (M20) were analyzed by using quantum theory of atoms in molecules (QTAIM) calculations and energy decomposition (EDA) techniques based on the orbitals optimized by density functional theory method (DFT). Imidazole (IMI) and carbene ligands (a-NHC and n-NHC) were considered for their interaction on the apex and face center position of the three different coinage metal clusters Au20, Ag20 and Cu20. The adsorption energies indicated the following behaviour in those complexes: (1) ligands adsorbed on the apex-A position of M20 clusters are more stable and less reactive, (2) the chemical stability of the carbene ligands is high, and (3) of the two NHCs considered here, our findings show a higher interfacial binding strength for a-NHC with M20 surfaces. Natural population analysis showed the charge transfer from imidazole to M20 with N–M coordination bonding and the existence of strong C–M covalent bonding for carbene-n,a-NHC–M20-complexes. QTAIM calculations again confirmed the covalent interactions in the latter complexes. Furthermore, energy decomposition analyses were performed to obtain the energetic properties of bonding for all the complexes.
1. Introduction
The interaction of metals with organic molecules has drawn enormous interest from chemical and materials science due to the fundamental physical and chemical phenomena involved.1 Indeed, compounds having a single metal atom and multiple organic molecules are well established in classical organometallic and coordination chemistry. However, recent technological progress and the desire for new applications generate a continuous demand for novel materials. More recently, the interaction of metal nanoparticles with biologically active molecules has become a substantial topic in biocatalysis, biocompatibility and biosensors.2 This has triggered an intensive research effort focusing on the electronic structure at the interface between biomolecules and inorganic materials. As a matter of fact, understanding the nature of the interaction between them is a not a trivial matter owing to the size and structural complexities of biomolecules like proteins or peptides.Due to these limitations, several studies were focused on the simpler entities like amino acids that are individual constituents of proteins and their adsorption on the metal surfaces provide a reasonable model to explain more complex biochemical interactions.3 Different experimental surface science techniques such as reflection-absorption infrared spectroscopy (RAIRS), photoelectron diffraction, XPS, and NEXAFS, along with theoretical approaches such as density functional theory (DFT), have been employed to study the adsorption of amino acids on gold and copper surfaces.4–15
Nevertheless, the various chemical groups, (the side chain, the N and C termini) that compose each amino acid, serve as the potential metal binding sites and a fundamental investigation on the interface between the amino acid functional group and the metal surface becomes necessary from both experimental and theoretical perspective. One such functional species of biological importance is imidazole (IMI), an interesting nitrogen heterocyclic molecule. It is a functional moiety of several biomolecules such as purine, nucleic acids, histidine and histamine.16 Imidazole and some of its derivatives have also been used as corrosion inhibitors and adhesion promoters, specifically for copper.17 Imidazole has two nitrogen atoms, labeled as N1 and N3, with N1 protonated while N3 deprotonated in its neutralized form (Fig. 1a). It most likely adsorbs on the metal surface through N3 atom owing to unshared its electron lone pair. This type of bonding is extensively demonstrated by the adsorption of many nitrogen heterocycles including pyridine on metal surface through the unshared electron lone pair of the nitrogen atom. A case study was reported recently with the DFT calculations on imidazole interaction with Au(111) surface.18 However a theoretical study of nature of bonding of the imidazole with copper and silver metal surfaces is not yet been fully elucidated and a detailed study is necessary.
 | ||
| Fig. 1 Schematic sketch of (a) IMI, (b) n-NHC and (c) a-NHC ligands. | ||
While nitrogen heterocycles like imidazole have a great history in biomedical research and still being a front-runner for bioactive applications, another class of nitrogen heterocycles that have recently become universal ligands in organometallic and inorganic coordination chemistry is N-Heterocyclic Carbenes (NHCs). The chemistry of NHCs has experienced significant development since 1991 when Arduengo et al. isolated the first stable NHC.19 NHC ligands usually coordinate to metals at C2 position (Fig. 1b), and until recently, only complexes that have “normal” NHC ligands (n-NHCs) were known. The first C4/C5 coordinated NHC complex, supposed to be “abnormal” carbene complex, was synthesized by Crabtree and co-workers20 and no rearrangement to the C2-bonded isomer has been observed, even under heating. There upon, several other complexes with “abnormal” carbene ligands (a-NHCs; Fig. 1c) have been synthesized by the same group21 and by others.22 NHCs are shown to be reasonable mimics of imidazole ligands and their steric and electronic properties are largely influenced by the substituents on the N atoms of the NHC ring, which plays a crucial role in the binding interactions between metal complexes and biomolecules. The first study on the biomedicinal applications of NHC ligands with metal [Rh(I) and Ru(II)] complexes on the antimicrobial activity have been published by Cetinkaya et al.23 A few years after this pioneering work, the groups of Berners-Price (2004) and Youngs (2005) with their works on the antitumor properties of Au(I)–NHC complexes and on the antimicrobial activity of Ag(I)–NHC complexes, respectively, initiated a number of studies in this research field.24,25 NHC is neutral when compared to anionic C deprotonated alkyl and aryl ligands. The cationic metal complexes can gain easier access to go across cell membrane whereas the neutral NHCs serve as excellent auxillary ligand in the construction of bioactive metal complexes. Besides, NHC ligand can be easily modified to allow for tuning of liphophility and among all NHCs, n-NHCs are currently the most widely explored carbene ligands used in biological studies. Fürstner and coworkers recently reported a general synthetic route to obtain substituted imidazolium, a precursor of NHC ligands, compounds.26 The substituent at two N, C4 and C5 atoms can be modified and thus modification at different interacting sites indeed provides useful active sites for tuning binding interactions of metal–NHC complexes with biomolecules. Apart from this, water soluble NHC–Cu complexes are now used as a catalysts and successful application of these complexes in bioconjugation using unprotected peptides acting as DNA binding domains was achieved very recently for the first time.27 Regardless of NHCs versatility in molecular coordination chemistry, its surface coordination chemistry has not been explored much. Just recently a few experimental investigations have been carried out on n-NHCs interacting with metal nanoparticles and n-NHC-based self assembled monolayers on gold substrate.28–30 Also there exist a recent study that describes the generation of N-heterocyclic carbene (NHC)-based SAMs on gold that demonstrate considerably greater resistance to heat and chemical reagents than the thiol-based counterparts.31 But a detailed theoretical exploration governing NHCs interaction with the metal surfaces, primarily necessary to tune the binding of metal–NHCs with biomolecules, is still lacking. Thus we are interested for such study, and among various NHCs, we have selected imidazol-2-ylidene (n-NHC) and imidazol-4-ylidene (a-NHC). The former has a peculiar stability and considered as a parent of all NHCs and for the latter no study is yet available related its interactions with metal surface/nanoparticles. As the selected NHCs are tautomers of imidazole, it is equally important to shed light on their interactions with such noble metals which aids in the designing of novel materials.
To achieve this, coinage metal surfaces namely gold (Au), silver (Ag) and copper (Cu) were chosen here as substrates for binding due to their catalytic, optical and electrical properties leading them contributing numerous applications in catalysis32,33 and optoelectronics.34–37 In fact, gold is considered to be the least reactive among the noble metals,38 while copper is with higher reactivity.5,11
Moreover, to understand the molecule–metal surface interfacial structure, electronic structure modelling on the atomic scale plays a crucial role as it provides key insights into the type and strength of the bonds between the molecule and the metal. Electronic structure methods are ideal tools but require high computational costs. For this reason, most electronic structure studies have adopted small metal clusters to mimic metal surfaces. Among small metal clusters, the tetrahedral 20-metal cluster (Td-20) is studied extensively for its nature of high symmetry. The tetrahedral geometry of a piece of fcc bulk metal is described with its twenty atoms on its surface and each of its four faces forming a plane (111). Thus, it has a very high surface area and a large fraction of corner sites with low coordination. The clusters within this size ranges are used in most industrial and academic application of catalysis. The three different kinds of atoms in the Td structure, 4 at the apexes, 4 at the centre of each face, and 12 along the edges (Fig. 2), have different coordination environments and may provide ideal surface sites to bind different molecules for catalysis (such as CO, O2, and CO2).39 It has been found that the most stable geometries of Au20 and Ag20 are tetrahedral Td structures while Cu20 preferred a compact structure with Cs symmetry.40 However, in this study, we have performed calculations on tetrahedral structures for all coinage metal clusters M20 (M = Au, Ag and Cu) at apical (A) and face centre (F) positions as we want to provide a small cluster model that mainly mimics the interactive site at metal(111) surface.
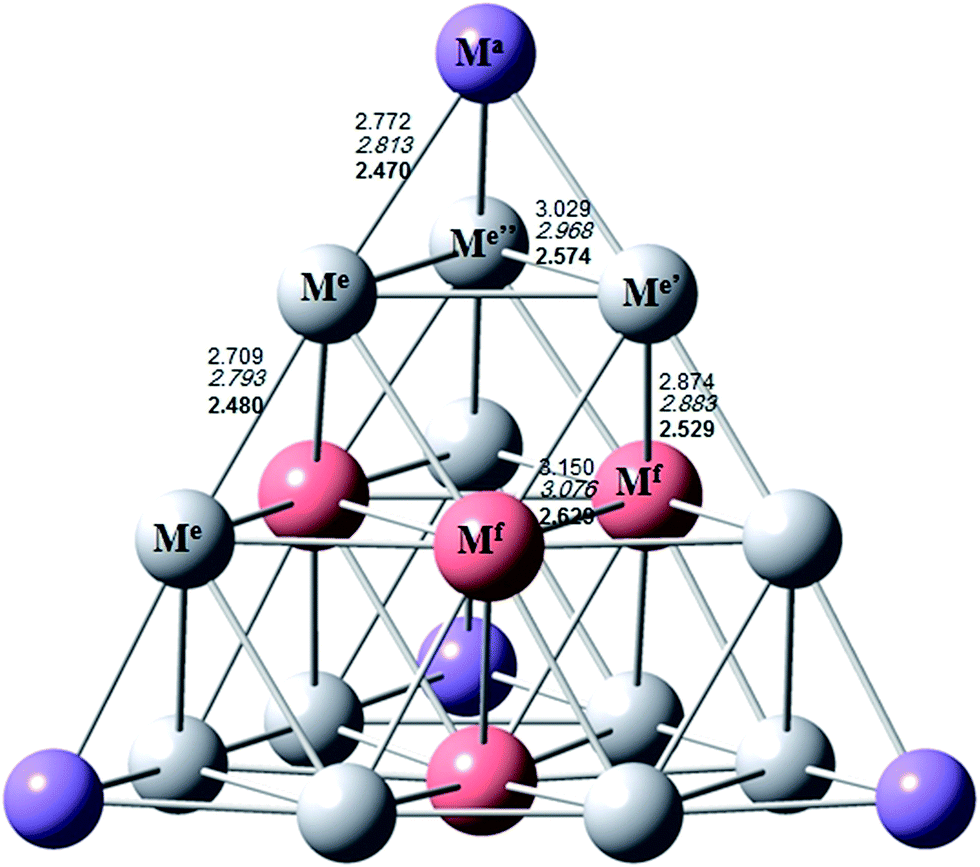 | ||
| Fig. 2 Bond distance of M20 (M = Au, Ag and Cu) clusters in Å (normal case for Au, italics for Ag and bold face for Cu) at the CAM-B3LYP/LANL2DZ level of theory. | ||
The interaction between six membered aromatic molecule like pyridine, containing heterogeneous atoms, and metallic clusters are investigated theoretically and the results show that the donation from the nitrogen lone-pair electrons to an unoccupied orbital plays a crucial role in the bonding.14,41,42 Recently, Prakash et al. reported the existence of the subtle competition between σ H-bond and π stacking interaction between CO2 and imidazole either isolated, adsorbed on a gold cluster or adsorbed on a gold surface.43 However, no investigation has been reported yet on five membered imidazole ring interacting with other noble metal clusters like silver and copper which intrigued this study. Here, we have studied imidazole with Au20 using CAM-B3LYP hybrid exchange correlation functional, different from PBE functional used by Prakash et al.43 For consistency we have chosen CAM-B3LYP hybrid functional for imidazole interactions with silver and copper clusters.
To the best of our knowledge, this is the first modelling study on the bonding behaviour of tautomers of imidazole (NHCs) with metal nanoparticles, particularly of size M20. As there exits a growing interests in combining nanoparticles for the detection of bio-molecules,44–48 biological imaging, and targeted drug delivery,44,49,50 with our electronic structure calculations and analyzing the nature and strength of respective M–N3, M–C2 and M–C4 bond at two different coordination sites in the IMI, n-NHC and a-NHC–metallic (M20, M = Au, Ag and Cu) cluster complexes, we hope to give a better understanding on the basic principle underlying the interaction of imidazole based ligands with metal clusters.
The remaining content of this paper is organized as follows. First we describe the details of our calculations. Next we discuss the results for the bare metal cluster and its complexation with IMI, n-NHC and a-NHC and geometries, binding energies and bonding nature.
2. Computational details
The calculations for the complexes analyzed here were performed with the Gaussian09 set of codes51 and the optimizations led to energetic minima since no imaginary frequencies were observed. A Coulomb-attenuated hybrid exchange-correlation functional (CAM-B3LYP)52 was applied with LANL2DZ relativistic effective core potentials and basis sets for metals (Au, Ag and Cu) and the Pople style 6-311G(d) basis sets for C, N and H atoms. This choice of the functional and basis sets seems to be justified since CAM-B3LYP has been earlier successfully applied in the calculations on gold clusters as well as of the numerous organic molecules while the LANL2DZ results are in agreement with the experimental data and the high level theoretical calculations.53,54For M20–ligand complexes, the geometry optimization was done with CAM-B3LYP/6-311G*(C, N, H)/LANL2DZ (Au, Ag, Cu) with default 5D. Hereafter, we represent the method and basis set just with CAM-B3LYP/6-311G*/LANL2DZ which denotes 6-311G* basis set for C, N, H, and LANL2DZ basis set for Au, Ag and Cu.
To explain the nature of bonds, Energy decomposition (EDA) and quantum theory of atoms in molecules (QTAIM) analyses were carried out on the CAM-B3LYP/6-311G*/LANL2DZ optimized structures. The EDA was done with the program package ADF version 2010.99 which is based on the work by Zigler and Rauk, and Morokuma.55,56 The bonding analysis was carried out using B3LYP/TZ2P level of theory and we considered the scalar relativistic effects using the zero-order regular approximation (ZORA).57 Further, we have used Voronoi Deformation Density (VDD) method implemented in ADF to deduce atom charges from the self-consistent results of a molecular calculation.58
The wave function data obtained in geometry optimization calculations were used to calculate the electron density of the complexes and perform the quantum theory of atoms-in-molecules (QTAIM) topology analysis,59,60 which focuses on molecular electron density rather than molecular orbitals. In the framework of QTAIM theory, three descriptors, i.e. the electron density ρBCP at the bond critical point (BCP), the Laplacian of electron density at BCP (∇2ρBCP), and the total energy density at BCP (HBCP), have been used to characterize the interaction between metal and the ligands.
In addition, we analyzed the CT properties between M20 clusters and IMI, n-a-NHCs using NBO61 approach. NBO analysis was carried out at CAM-B3LYP/6-311G*/LANL2DZ level of theory.
3. Results and discussion
3.1 Geometries of M20 (M = Au, Ag and Cu) clusters, imidazole and its tautomers
Fig. 2 summarizes the calculated CAM-B3LYP/LANL2DZ level of theory results of the optimized geometries of gold, silver and copper clusters (M20 clusters). The geometry of the tetrahedral M20 cluster is represented by five distinct M–M bond distances between the three non-equivalent sites: apex-Ma, edge-Me, and face center-Mf (see Fig. 2). The Aua–Aue, Aue–Aue, Aue′–Aue′′, Aue–Auf and Auf–Auf bond distances are equal to 2.772, 2.709, 3.029, 2.874 and 3.150 Å, respectively. Regarding Ag20 and Cu20, the respective bond distances are shown in italics and bold-face numbers in the Fig. 2. The bond distances between the copper atoms are the shortest among three metal clusters considered here and in particular the Cua–Cue bond distance (2.470 Å) between the apex and edge copper atoms is the shortest of all.The optimized geometries of the free imidazole isomers IMI, n-NHC, and a-NHC calculated at CAM-B3LYP/6-311G* level are shown in Fig. 3, along with the most important bond lengths and angles and the relative energies of the ligands. The C–N–C, N–C–N and N–C–C angles for imidazole and n,a-NHCs are 105.3, 100.7 and 100.1°, respectively. We have found that our calculated relative energies IMI (0.0 kcal mol−1) < n-NHC (26.4 kcal mol−1) < a-NHC (47.5 kcal mol−1) are in good agreement with the previous available theoretical data.62
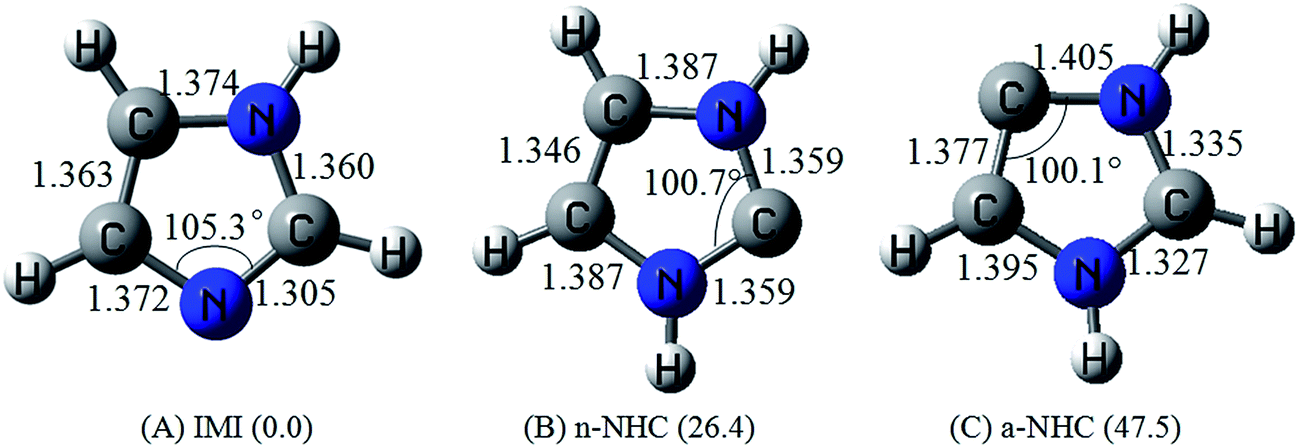 | ||
| Fig. 3 Calculated geometries of the ligands (bond lengths in Å, angle in degree) at the CAM-B3LYP/6-311G* level of theory. Relative energies (in kcal mol−1) are given in parentheses. | ||
3.2 Geometries of IMI–M20 and n-NHC, a-NHC–M20
For our calculations, we have chosen two different active binding sites (Ma and Mf hereafter A and F positions) at non-equivalent coordination sites of the metal (M20) clusters as shown in the Fig. 2. The ground state Cartesian coordinates and the calculated energies are given in SI. The geometries of the IMI–M20, n-NHC–M20 and a-NHC–M20 complexes at binding site A and F are listed in Table 1. They reveal that there are weak and strong interactions between the chosen ligands (IMI, n-NHC and a-NHC) and the neutral metal (M20) clusters.| IMI-complexes | M20 | A | F | n-NHC complexes | A | F | a-NHC complexes | A | F |
|---|---|---|---|---|---|---|---|---|---|
| N1–C2 | Au | 1.347 | 1.350 | N1–C2 | 1.346 | 1.346 | N1–C2 | 1.327 | 1.329 |
| Ag | 1.350 | 1.353 | 1.348 | 1.346 | 1.326 | 1.328 | |||
| Cu | 1.347 | 1.350 | 1.349 | 1.349 | 1.327 | 1.328 | |||
| C2–N3 | Au | 1.313 | 1.310 | C2–N3 | 1.346 | 1.340 | C2–N3 | 1.329 | 1.324 |
| Ag | 1.311 | 1.308 | 1.348 | 1.384 | 1.330 | 1.328 | |||
| Cu | 1.313 | 1.310 | 1.349 | 1.349 | 1.329 | 1.327 | |||
| N3–C4 | Au | 1.375 | 1.373 | N3–C4 | 1.383 | 1.384 | N3–C4 | 1.394 | 1.390 |
| Ag | 1.375 | 1.373 | 1.383 | 1.384 | 1.396 | 1.392 | |||
| Cu | 1.376 | 1.374 | 1.383 | 1.384 | 1.398 | 1.396 | |||
| C4–C5 | Au | 1.358 | 1.360 | C4–C5 | 1.347 | 1.348 | C4–C5 | 1.367 | 1.365 |
| Ag | 1.360 | 1.361 | 1.346 | 1.347 | 1.368 | 1.368 | |||
| Cu | 1.359 | 1.360 | 1.347 | 1.348 | 1.369 | 1.369 | |||
| N1–C5 | Au | 1.375 | 1.376 | N1–C5 | 1.383 | 1.380 | C5–N1 | 1.386 | 1.387 |
| Ag | 1.375 | 1.375 | 1.384 | 1.385 | 1.388 | 1.389 | |||
| Cu | 1.376 | 1.376 | 1.383 | 1.383 | 1.388 | 1.389 | |||
| N3–M20 | Au | 2.235 | 2.348 | C2–M20 | 2.103 | 2.037 | C4–M20 | 2.098 | 2.035 |
| Ag | 2.330 | 2.470 | 2.258 | 2.296 | 2.236 | 2.259 | |||
| Cu | 2.032 | 2.116 | 2.011 | 2.031 | 2.004 | 2.027 | |||
| C2–N3–M | Au | 125.73 | 125.07 | N1–C2–M | 128.39 | 130.01 | C5–C4–M | 133.88 | 135.71 |
| Ag | 126.05 | 125.60 | 127.77 | 124.85 | 134.01 | 138.35 | |||
| Cu | 125.86 | 126.26 | 129.49 | 129.61 | 134.54 | 136.71 | |||
| C4–N3–M | Au | 127.52 | 128.24 | N3–C2–M | 128.13 | 125.83 | N3–C4–M | 123.17 | 120.50 |
| Ag | 127.64 | 128.19 | 129.51 | 132.32 | 123.73 | 119.23 | |||
| Cu | 127.67 | 127.31 | 127.73 | 127.53 | 123.24 | 120.93 | |||
| R.E | Au | 0.0 | 0.0 | R.E | −9.12 | −4.79 | R.E | −22.22 | −16.09 |
| Ag | 0.0 | 0.0 | −17.56 | −17.83 | −32.31 | −33.35 | |||
| Cu | 0.0 | 0.0 | −16.75 | −14.90 | −31.35 | −30.95 | |||
| B.E | Au | −19.72 | −8.72 | B.E | −37.23 | −19.15 | B.E | −45.26 | −40.34 |
| Ag | −15.08 | −6.80 | −24.01 | −15.49 | −30.42 | −21.08 | |||
| Cu | −23.75 | −12.35 | −33.52 | −23.98 | −40.04 | −29.04 | |||
| ΔG | Au | −11.83 | 0.71 | ΔG | −29.18 | −21.83 | ΔG | −36.46 | −31.45 |
| Ag | −7.73 | 0.16 | −17.75 | −7.49 | −22.42 | −12.62 | |||
| Cu | −16.0 | −3.95 | −25.82 | −15.62 | −31.65 | −20.38 |
Let us now concentrate on the M–N, M–C2 and M–C5 bond distances at the active site. For the same metal as substrate, we see that the respective bond distances are shorter at the A sites whereas at F position they are longer. For the Au–N, the short bond distance of 2.235 Å at the atom binding site A and the large bond distance of 2.348 Å at the site F representing (111) surface are observed. The latter agrees well with the GGA/PBE optimized imidazole/Au(111) interface characterized by a Au–N distance of ≈2.3 Å.63,43 In all these cases there is no direct experimental data for comparison. For Cu–N the bond distances are within the range of 2.023–2.116 Å, which are close to 2.00 (±0.02) Å obtained by using the photoelectron diffraction technique for pyridine adsorbed on Cu(110).64
For the identical metal clusters bound with the different ligands the sequence of relative energy of the complexes are a-NHC–M20 > n-NHC–M20 > IMI–M20 for all three kinds of metal clusters as shown in Table 1. Also, just at a first glance on binding energy columns of Table 1 one can notice that the ligand adsorbed on the apex position-A is more stable than on the F positions in all M20 systems. In addition, the C–N and C–C bond distances within the ring of the ligands do not show much variation among the different metal substrates considered here for binding. However, there exists significant energetic and the M–M bonding differences in the metal cluster complexes at each coordination sites indicating the strength of binding interactions. Among IMI–M20 complexes; for IMI–Cu20 at apex position the Cua–Cua bond becomes 0.039 Å longer after its binding when compared to bare metal cluster Cua–Cua (1.377 Å), whereas its N–Cu bond is the shortest (2.032 Å) owing to strong binding. But on the F binding site, N–Cu bond is 0.074–0.084 Å longer compared to A site thus showing weaker binding. The trend in the change of the M–M bond distances can also be found in the IMI–Au20 and IMI–Ag20 systems. We should note that the changes are considerable. It can be understood that the interactions are stronger in the bonding between the imidazole and the Cu20 clusters than the corresponding ligand interactions with Au20, Ag20 clusters. The shortest change of 0.026 Å in Aga–Aga bond distance indicates the weak binding of Ag–M20 (2.258 Å).
Nevertheless, one can notice that the order of the binding strength changes when n-NHC and a-NHC interacts with the M20 clusters. The C–M bond distances for the n-NHC on Au cluster is 2.103 and 2.037 Å at the A and F positions, slightly shorter than the recently determined theoretical value 2.118 Å for the n-NHC based ligands on Au(111) surfaces.31 Though C2–Cu and C4–Cu bonds are the shortest in the table, the binding energy for the n-NHC, a-NHC–Au20 systems shows higher. This is due to the fact that Aua–Aua bond distances in the complexes are distorted and shows longer than the bare Au20 cluster. Detailed analyses and explanation of the nature of the metal–ligand bonding at the active site are given in the following sections. Apart from relative energy and binding energy, Table 1 show the change in Gibbs free energy (ΔG) for all the M20–ligand complexes. The sequences of ΔG follow the similar trend as that of binding energy. However, more negative values for ΔG are observed for all the complexes at A positions correlating the contribution of entropy favors the complex formations.
3.3 QTAIM and energy decomposition analysis
BCP corresponds to the minimum of the electron density along the bond path of an atom pair, with which the chemical bonding interactions may be characterized according to the properties of electron and energy densities. According to Bader,59,60 based on the analysis of various systems, a ρBCP value of 0.2 e− per bohr3 may work as the lower bound to judge a covalent bond and a value of 0.1 e− per bohr3 as an upper bound for closed-shell interactions including ionic interactions. Meanwhile, the values of ∇2ρ < 0 and H < 0 refer to the shared interaction or covalent bond, while the values of ∇2ρ > 0 and H > 0 are indicators of ionic or hydrogen bonds and van der Waals interactions.59,60The corresponding QTAIM topology analysis data for the metal–N/C bond critical points in all complexes studied are summarized in Table 2, in which all indexes show the similar trend with the electron density ρ. Fig. 4 illustrates the electron density at BCP for all the cases, indicating that the metal–N/C bond is mainly ionic due to minor electron accumulations between the metals and the ligands with ρBCP value smaller than 0.13 e− per bohr3 calculated for all complexes. Nevertheless, it is noticed that the ρBCP values of Au–C bonds in the complexes with n,a-NHC ligands adsorbed on Au cluster are larger than other complexes (0.11–0.13 and 0.04–0.09 e− per bohr3 for the complexes of n,a-NHC adsorbed on Au cluster and other complexes, respectively), suggesting a stronger covalency existed in the Au–C bonds for the absorption of n,a-NHC ligands on Au cluster. In addition, the trend of the electron density ρBCP is well consistent with that of bonding energy, indicating that the covalent character in ionic bond plays an important role for the absorption capability, i.e. the larger covalent contribution to ionic bonds, the stronger interaction for the absorption between the metal clusters and ligands. We note that there is moderately larger build-up of electron density between n,a-NHC and M20 clusters than between IMI and the metal clusters, stemming from the electron donor of active carbene. The last column in Table 2 show the calculated AIM charge for the ligands in the complexes and it is basis set dependent.
| Complexes | Positions | BCPa | ρBCP (a.u) | ∇2ρBCP (a.u) | VBCP (a.u) | GBCP (a.u) | HBCP (a.u) | AIM charge for ligands |
|---|---|---|---|---|---|---|---|---|
| a Atomic numbering refers to Fig. 2. The electron density (ρBCP), Laplacian of the electron density (∇2ρBCP), potential energy density (VBCP), kinetic energy density (GBCP), and energy density (HBCP) at the BCP, respectively. The last column denotes the calculated AIM charges for the ligands in the complexes. | ||||||||
| IMI–Au20 | A | N–Au | 0.0764 | 0.3000 | −0.1051 | 0.0900 | −0.0151 | 0.16 |
| F | N–Au | 0.0615 | 0.2301 | −0.0789 | 0.0682 | −0.0107 | 0.12 | |
| IMI–Ag20 | A | N–Ag | 0.0546 | 0.2403 | −0.0728 | 0.0664 | −0.0064 | 0.10 |
| F | N–Ag | 0.0408 | 0.1767 | −0.0477 | 0.0459 | −0.0018 | 0.03 | |
| IMI–Cu20 | A | N–Cu | 0.0788 | 0.4473 | −0.1183 | 0.1150 | −0.0033 | 0.08 |
| F | N–Cu | 0.0607 | 0.3622 | −0.0873 | 0.0889 | −0.0355 | 0.05 | |
| n-NHC–Au20 | A | C–Au | 0.1111 | 0.3126 | −0.1490 | 0.1135 | −0.0355 | 0.30 |
| F | C–Au | 0.1288 | 0.3364 | −0.1813 | 0.1825 | 0.0012 | 0.34 | |
| H–Au | 0.0145 | 0.0458 | −0.0103 | 0.0108 | 0.0005 | |||
| n-NHC–Ag20 | A | C–Ag | 0.0716 | 0.2341 | −0.0919 | 0.0752 | −0.0167 | 0.24 |
| F | C–Ag | 0.0662 | 0.2199 | −0.0881 | 0.0690 | −0.0191 | 0.17 | |
| n-NHC–Cu20 | A | C–Cu | 0.0872 | 0.3954 | −0.1264 | 0.1126 | −0.0138 | 0.15 |
| F | C–Cu | 0.0832 | 0.3790 | −0.1176 | 0.1062 | −0.0114 | 0.13 | |
| a-NHC–Au20 | A | C–Au | 0.1126 | 0.2943 | −0.1473 | 0.1103 | −0.0370 | 0.37 |
| F | C–Au | 0.1288 | 0.3088 | −0.1701 | 0.1231 | −0.0470 | 0.39 | |
| H–Au | 0.0179 | 0.0518 | −0.0113 | 0.0121 | 0.0008 | |||
| a-NHC–Ag20 | A | C–Ag | 0.0752 | 0.2843 | −0.0959 | 0.0772 | −0.0187 | 0.25 |
| F | C–Ag | 0.0719 | 0.2249 | −0.0903 | 0.0732 | −0.0171 | 0.21 | |
| H–Ag | 0.0073 | 0.0142 | −0.0031 | 0.0033 | 0.0002 | |||
| a-NHC–Cu20 | A | C–Cu | 0.0890 | 0.3855 | −0.1256 | 0.1114 | −0.0142 | 0.20 |
| F | C–Cu | 0.0848 | 0.3661 | −0.1169 | 0.1042 | −0.0127 | 0.15 | |
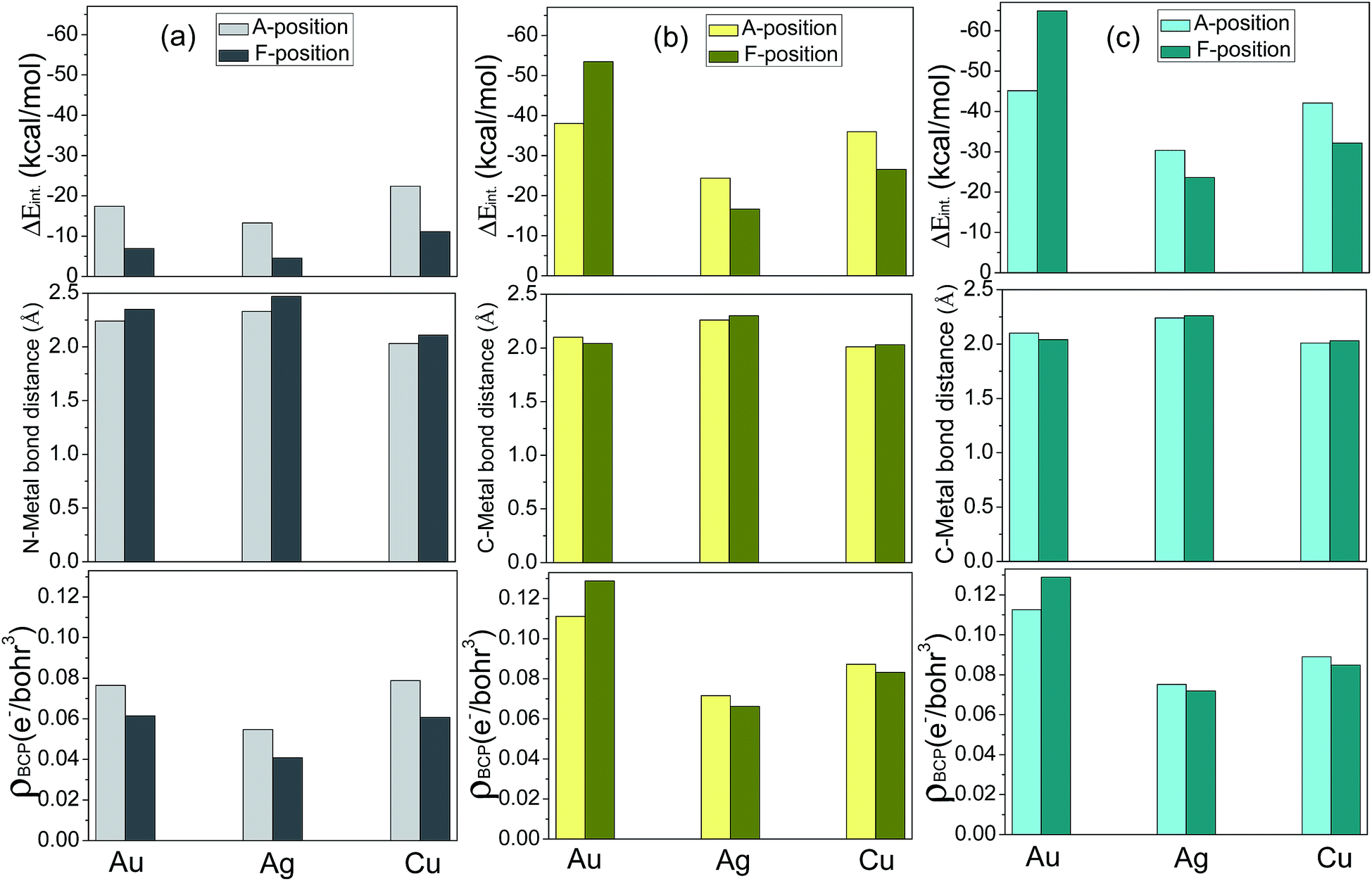 | ||
| Fig. 4 The calculated binding energy (top), bond distance between active site and the metal (middle), and electron density at the BCP (bottom) for the metal M20 complexes with the ligands (a) imidazole (IMI) (gray), (b) n-NHC (yellow), and (c) a-NHC (cyan). Light and dark color for A and F coordination modes, respectively. | ||
With QTAIM analysis, precious insight is obtained into the bonding situation of the M–N, M–C1 and M–C5 bond in the IMI, n-NHC, a-NHC–M20 complexes at A and F positions. Besides QTAIM analysis, the chemical bond between the two fragments can also be analyzed by examining the redistribution of the charge density when forming the complexes by using Voronoi deformation density (VDD)58 method in ADF. The VDD method does not explicitly use the basis functions, but calculates the amount of electronic charge density entering or leaving a certain atom due to bond formation by spatial integration of the deformation density over the atomic Voronoi cell. From the calculated values of VDD charge in Table 4, we find that for IMI, n,a-NHC–Au20 complexes, the charge flow is maximum from IMI, n,a-NHC ligands to Au20 at A species when compared to F species. For rest of the metal clusters there is not much differences in charge flowing from ligands to metal clusters between the two species, A and F positions. However, from the calculated AIM charges in Table 2 one can infer just opposite trend from VDD where the flow of charge from the ligands is maximum at F positions for n,a-NHC–Au20 complexes.
Concerning the nature of chemical bonding between the M20–ligand complexes, explorations were done by examining the energetic properties of the bonding by employing EDA. In this method, the total bonding energy between two or more fragments, ΔEint, consists of three physically meaningful components:
| ΔEint = ΔEelstat + ΔEPauli + ΔEorb |
| Complexes | Site | ΔEelstat | ΔEPauli | ΔEorb | ΔEint | qVDD(L) | qNPA(L) | qAIM(L) |
|---|---|---|---|---|---|---|---|---|
| IMI–Au20 | A | −78.09 (70.25) | 93.77 | −33.07 (29.75) | −17.39 | 0.21 | 0.14 | 0.16 |
| F | −56.86 (69.24) | 75.19 | −25.27 (30.77) | −6.93 | 0.19 | 0.19 | 0.12 | |
| IMI–Ag20 | A | −48.64 (73.15) | 53.24 | −17.85 (26.85) | −13.25 | 0.15 | 0.09 | 0.10 |
| F | −33.20 (70.62) | 42.45 | −13.81 (29.38) | −4.56 | 0.15 | 0.15 | 0.03 | |
| IMI–Cu20 | A | −72.61 (71.62) | 78.99 | −28.77 (28.38) | −22.39 | 0.17 | 0.11 | 0.08 |
| F | −59.47 (69.35) | 74.66 | −26.28 (30.65) | −11.09 | 0.18 | 0.18 | 0.05 | |
| n-NHC–Au20 | A | −173.73 (73.71) | 197.67 | −61.95 (26.29) | −38.02 | 0.24 | 0.27 | 0.30 |
| F | −209.00 (72.64) | 234.28 | −78.81 (27.39) | −53.43 | 0.21 | 0.36 | 0.34 | |
| n-NHC–Ag20 | A | −90.95 (75.99) | 95.34 | −28.74 (24.01) | −24.35 | 0.16 | 0.18 | 0.24 |
| F | −84.00 (73.36) | 97.87 | −30.51 (26.64) | −16.64 | 0.17 | 0.24 | 0.17 | |
| n-NHC–Cu20 | A | −111.81 (74.21) | 114.71 | −38.84 (25.78) | −35.95 | 0.17 | 0.20 | 0.15 |
| F | −110.56 (71.50) | 128.08 | −44.06 (28.49) | −26.54 | 0.18 | 0.28 | 0.13 | |
| a-NHC–Au20 | A | −183.03 (73.34) | 204.45 | −65.75 (26.35) | −45.12 | 0.27 | 0.28 | 0.37 |
| F | −220.09 (72.23) | 239.82 | −84.61 (27.77) | −64.88 | 0.23 | 0.36 | 0.39 | |
| a-NHC–Ag20 | A | −101.77 (75.66) | 104.17 | −32.74 (24.34) | −30.34 | 0.20 | 0.19 | 0.25 |
| F | −98.79 (72.85) | 112.01 | −36.82 (27.15) | −23.60 | 0.18 | 0.24 | 0.21 | |
| a-NHC–Cu20 | A | −119.06 (73.84) | 119.20 | −42.19 (26.16) | −42.05 | 0.19 | 0.20 | 0.20 |
| F | −116.70 (70.66) | 133.00 | −48.46 (29.34) | −32.15 | 0.20 | 0.29 | 0.15 |
As seen in Table 3, the interaction energy ΔEint receives an important part of its stabilizing character from the electrostatic interactive term ΔEelstat which contributes more to the total attractive interactions. The character of the bonds is almost ionic, however the absolute values of the energy terms ΔEPauli, ΔEelstat and ΔEorb reveal the details about the bonding situation. As we can see that each of the calculated values ΔEelstat and ΔEorb is much higher than the bonding energy, ΔEint. It is important to emphasize that an arbitrary consideration of just one attractive term, i.e., imidazole and carbene ligand is either only electrostatically bound or is only covalently bound cannot explain the physical reality of binding.
A complete examination of all the three terms gives the clear picture of binding. We note that for all the complexes, the electrostatic contribution is not large enough to compensate for Pauli repulsion and without the energy contribution from the orbital interaction the total interactive bonding energy would go repulsive. The electrostatic contribution for M–N and M–C bonding is higher (ranges between 70.25–75.66%) for apex-centered A-complexes. Thus the binding energy is stronger for the A positioned complexes which are in good agreement with their smaller M–N and M–C bonds at those positions. However, the covalent contribution is higher (ranges between 26.16–30.77%) for the F positioned complexes. The electrostatic and covalent character of the respective M–N and M–C bonding in the complexes decrease in the order of Au > Cu > Ag. Furthermore, one can observe that the absolute value of ΔEPauli, ΔEelstat and ΔEorb for the n,a-NHC–M20 complexes are larger than the IMI–M20 complexes.
Fig. 4 shows the calculated bonding energy from the two fragments with EDA scheme, bond distance between active site and the metal, and electron density at the BCP for the metal M20 complexes with the ligands (a) imidazole (IMI) (b) n-NHC (c) a-NHC. From the bonding energies, it can be concluded as follows: (1) complexes with three ligands adsorbed on the apex position-A are more stable than those on the face centered-F positions except for the cases of ligands n-NHC and a-NHC adsorbed on Au, in which the face-centered F-complexes with n,a-NHC ligands bound on Au show the binding superiority relative to the A-position ones. (2) The sequence of binding strength for identical ligand adsorbed on the different metal clusters is Cu > Au > Ag for IMI ligand and Au > Cu > Ag for n,a-NHC ligands, respectively. (3) The sequence of binding strength for the identical metal clusters bound with the different ligands is a-NHC > n-NHC > IMI for all three kinds of metal clusters. The range of bonding energy from the EDA scheme is −23.60 to −64.88/−16.64 to −53.43/−4.56 to −22.39 kcal mol−1 for the adsorption of ligand a-NHC/n-NHC/IMI, respectively. These three features can be reflected on the geometrical structures, especially for bond distance between active site and the metal. It can be found in Fig. 4 that the larger binding strength between the ligand and the metal cluster, the relative shorter bond distance between active site and the metal. In addition, we can see from Table 2 that the electron densities at the BCPs of the bonds between the metals and the ligands are consistent with these above features.
3.4 Molecular orbital and NBO analysis
Energies and shapes of frontier molecular orbitals HOMO and LUMO, as well as the HOMO–LUMO energy gaps, are shown in Fig. 5. It can be noticed that both interacting atoms of ligands and Au cluster have direct contribution to frontier molecular orbital HOMO in the interaction between Au cluster and three types of ligands, implying a favorable absorbability of ligands on the surface of Au cluster relative to other two metal clusters. In addition to this, in complexes with n,a-NHC adsorbed on three types of metal clusters, the orbital densities of HOMO are also placed on the metal and carbene C atom, while HOMOs of complexes of IMI with Ag and Cu clusters are mainly concentrated on metal clusters, suggesting an active behavior of n,a-NHC ligands relative to IMI ligand. By comparing the values of EHOMO for all complexes, it can be found that the complexes of ligands adsorbed on the Au cluster show lower HOMO energies (−6.15 to −6.75 eV and −5.25 to −5.63 eV for EHOMO of Au complexes and other metal complexes, respectively), especially for the adsorption at F position (EHOMO: −6.48 to −6.75 eV), indicating that it is difficult for Au complexes to be oxidized by losing electrons relative to other metal complexes. According to the energy gap between the HOMO and LUMO listed in Fig. 5, the interaction of ligand with metal cluster decrease their reactivities and probably increase their chemical stabilities following the order Au > Cu > Ag (ΔEg: 4.25–4.54, 3.97–4.14, and 3.95–4.08 eV for Au, Cu, and Ag complexes, respectively), in which Au complexes possess the lowest the reactivity, i.e. the highest chemical stability. Similarly, for the complexes with the different ligands adsorbed on same metal cluster, chemical stability follows the order IMI > n-NHC > a-NHC according to the values of ΔEg (4.06–4.54, 3.97–4.33, and 3.95–4.28 eV for IMI, n-NHC, and a-NHC, respectively), in which the complexes with n,a-NHC show high reactivity relative to the complexes with IMI. | ||
| Fig. 5 Shapes of the frontier molecular orbitals HOMO and LUMO, energies of HOMO and LUMO, and HOMO–LUMO energy gaps (ΔEg) for complexes (energies in eV). | ||
A second-order perturbation theory analysis of the Fock matrix was also carried out to evaluate the donor–acceptor interaction on the NBO basis. In Table 4, the perturbative stabilization energies (ΔECT) for the M–N and M–C bonds in IMI–M20 and a,n-NHC–M20 complexes are given.
| Complexes | Positions | Charge transfera | ΔECT | NPA charges for ligands |
|---|---|---|---|---|
| a σnb – non-bonded, σ*nb – unfilled non-bonded orbital that receives charge from lone pair nitrogen, σ* – anti-bonding orbital. | ||||
| IMI–Au20 | A | σnbN21 → σ*nbAu13 | 45.32 | 0.14 |
| F | σnbN21 → σ*nbAu4 | 29.78 | 0.19 | |
| IMI–Ag20 | A | σnbN21 → σ*nbAg13 | 17.40 | 0.09 |
| F | σnbN21 → σ*nbAg4 | 26.17 | 0.15 | |
| IMI–Cu20 | A | σnbN21 → σ*nbCu13 | 28.61 | 0.11 |
| F | σnbN21 → σ*nbCu4 | 48.42 | 0.18 | |
| n-NHC–Au20 | A | σnbAu13 (α) → σ*C13–N24 | 2.67 | 0.27 |
| (β) → σ*C13–N26 | 10.15 | |||
| F | σnbAu4 (α) → σ*C28–N23 | 3.50 | 0.36 | |
| (β) → σ*C28–N24 | 3.37 | |||
| n-NHC–Ag20 | A | σnbAg13 (α) → σ*C13–N24 | 1.30 | 0.18 |
| (β) → σ*C13–N25 | 3.86 | |||
| F | σnbAg4 (α) → σ*C24–N26 | 0.78 | 0.24 | |
| (β) → σ*C24–N25 | 3.13 | |||
| n-NHC–Cu20 | A | σnbCu13 (α) → σ*C13–N24 | 1.15 | 0.20 |
| (β) → σ*C13–N25 | 6.21 | |||
| F | σnbCu4 (α) → σ*C25–N23 | 0.82 | 0.28 | |
| (β) → σ*C25–N24 | 5.12 | |||
| a-NHC–Au20 | A | σnbAu13 (α) → σ*C27–C21 | 7.70 | 0.28 |
| (β) → σ*C27–N28 | 3.01 | |||
| F | σnbAu4 (α) → σ*C27–N28 | 3.99 | 0.36 | |
| (β) → σ*C27–C21 | 9.13 | |||
| a-NHC–Ag20 | A | σnbAg13 (α) → σ*C27–C21 | 3.28 | 0.19 |
| (β) → σ*C27–N28 | 1.24 | |||
| F | σnbAg4 (α) → σ*C27–N28 | 2.95 | 0.24 | |
| (β) → σ*C27–C21 | 0.98 | |||
| a-NHC–Cu20 | A | σnbCu13 (α) → σ*C27–N28 | 1.24 | 0.20 |
| (β) → σ*C27–C21 | 4.66 | |||
| F | σnbCu4 (α) → σ*C27–N28 | 1.24 | 0.29 | |
| (β) → σ*C27–C21 | 3.61 | |||
In the case of IMI–M20 (M = Au, Ag and Cu) complexes at A and F positions, charge is transferred from the lone pair (LP) of the nitrogen atom to non-occupied non-bonding orbital (σnb) of metal clusters. Thus, the interaction is the Lewis acid–base interaction with N–M coordination bonding, where nitrogen and metal atom act as Lewis acid and base, respectively. A comparison of M–N bonds from Table 1 demonstrates Cu–N is stronger than that of the other two metal complexes. Accordingly, total charge transfer energies, electron density values at BCPs are more than those IMI–Au20 and IMI–Ag20 metal complexes.
For n,a-NHC–M20 (M = Au, Ag and Cu) complexes at A and F positions, a different scenario is observed compared to IMI–M complexes. The charge is transferred from metal clusters to anti-bonding (σ*) orbitals of C–C and C–N in n,a-NHCs with covalent C–M bonding. There occurs stronger bond at the active site in the a,n NHCs–Au20 complexes than the other two metal complexes because of more stabilization energies associated with such bonding. Therefore, more ΔECT is observed in these Au metal complexes. The last column of Table 4 shows the calculated Natural Population Analysis (NPA) charge for the ligands and the trend goes in harmony with the AIM charge where here also the maximum charge flow from the ligand is observed for the F species but all the metal clusters are falling in the domain of charge acceptors which is quite different from VDD and AIM charges for the ligands.
4. Conclusion
In this work, density functional theory (DFT), QTAIM, and EDA calculations were employed to study the adsorption of imidazole based ligands on metal clusters M20. Two different positions (A and F) on M20 clusters were considered to study the adsorption. The adsorption energies indicated that ligands adsorbed on the apex-A position of M20 clusters are more stable. The sequence of binding strength for identical ligand adsorbed on the different metal clusters is Cu > Au > Ag for IMI ligand and Au > Cu > Ag for n,a-NHC ligands, respectively. The sequence of binding strength for the identical metal clusters bound with the different ligands is a-NHC > n-NHC > IMI for all three kinds of metal clusters. Natural population analysis showed the charge transfer from imidazole to M20 with N–M coordination bonding whereas the existence of a strong C–M covalent bonding for n,a-NHC–M20 complexes. Subsequent QTAIM calculations confirmed the covalent interactions in n,a-NHC–M20 complexes. Moreover, energy decomposition analyses showed high electrostatic contributions at A position and covalent contributions at F position for M–N and M–C bonding. For Cu–N(IMI) our calculated bond distances at A and F position are 2.023 and 2.116 Å, respectively, which are close to 2.00 (±0.02) Å obtained from the photoelectron diffraction technique for pyridine adsorbed on Cu(110).64 Finally, our studies indicate a high stability of IMI and Cu20 species that may serve for various exciting applications in the field of molecular electronic devices and energy materials apart from the common nitrogen based molecular interactions on gold nanoparticle/surfaces. The determination of a strong covalent bonds between n,a-NHCs and Au20 species from our computations may assist for self-assembled monolayers (SAMs) of organic molecules on gold metals and further applications of SAMs65 instead of the well-known gold–thiol, nitrogen counterparts. The experimental study on the SAMs of a-NHCs on the metal surfaces is not yet available. Among the two NHCs considered here, our findings show higher interfacial binding strength for a-NHC with M20 surfaces. Thus, our fundamental studies, on unravelling the nature of interfacial binding of the imidazole and imidazole-based NHCs ligands with coinage metal clusters, will certainly aid in mimicking imidazole with n,a-NHCs at biomolecule/metal interfaces thus opening a new avenue to generate new materials and molecular devices.Acknowledgements
K. R. G acknowledges the support by CSIC under JAE-DOC program 'Junta para la Ampliación de Estudios' cofinanciada por el FSE, and in part, by the Basque Departamento de Educación, Universidades e Investigación, the University of the Basque Country UPV/EHU (Grant No. IT-366-07), the Spanish Ministerio de Ciencia e Innovación (Grant No. FIS2010-19609-C02-02) and also the computational resources from DIPC and CSIC – Spain, during her stay over there.References
- J. Kuchar and R. P. Hausinger, Chem. Rev., 2004, 104, 509 CrossRef CAS PubMed.
- R. W. Murray, Chem. Rev., 2008, 108, 2688 CrossRef CAS PubMed.
- K. Ray, M. H. Chowdhury, H. Szmacinski and J. R. Lakowicz, J. Phys. Chem. C, 2008, 112, 17957 CAS.
- G. Xue, J. Dong and Y. Sun, Langmuir, 1994, 10, 1477 CrossRef CAS.
- B. Liedberg, C. Carlsson and I. Lundström, J. Colloid Interface Sci., 1987, 120, 64 CrossRef CAS.
- J. Hasselström, O. Karis, M. Weinelt, N. Wassdahl, A. Nilsson, M. Nyberg, L. G. M. Pettersson, M. G. Samant and J. Stöhr, Surf. Sci., 1998, 407, 221 CrossRef.
- M. Nyberg, J. Hasselström, O. Karis, N. Wassdahl, M. Weinelt, A. Nilsson and L. G. M. Pettersson, J. Chem. Phys., 2000, 112, 5420 CrossRef CAS PubMed.
- G. Dodero, L. de Michieli, O. Cavalleri, R. Rolandi, L. Oliveri, A. Dacca and R. Parodi, Colloids Surf., A, 2000, 175, 121 CrossRef CAS.
- S. M. Barlow and R. Raval, Surf. Sci. Rep., 2003, 50, 201 CrossRef CAS.
- M. E. Marti, Ch. Methivier, P. Dubot and C. M. Pradier, J. Phys. Chem. B, 2003, 107, 10785 CrossRef.
- M. E. Marti, A. Quash, Ch. Methivier, P. Dubot and C. M. Pradier, Colloids Surf., A, 2004, 249, 85 CrossRef PubMed.
- Y. Zubavichus, M. Zharnikov, Y. Yang, O. Fuchs, C. Heske, E. Umbach, G. Tzvetkov, F. P. Netzer and M. Grunze, J. Phys. Chem. B, 2005, 109, 884 CrossRef CAS PubMed.
- J. Jones, L. B. Jones, F. Thibault-Starzyk, E. A. Seddon, R. Raval, S. J. Jenkins and G. Held, Surf. Sci., 2006, 600, 1924 CrossRef PubMed.
- F. Iori, S. Corni and R. D. Felice, J. Phys. Chem. C, 2008, 112, 13540 CAS.
- V. Feyer, O. Plekan, T. Skála, V. Cháb, V. Matolin and K. C. Prince, J. Phys. Chem. B, 2008, 112, 13655 CrossRef CAS PubMed.
- J. M. Kogot, H. J. England, G. F. Strouse and T. M. Logan, J. Am. Chem. Soc., 2008, 130, 16156 CrossRef CAS PubMed.
- G. Bhargava, T. A. Ramanarayanan and S. L. Bernasek, Langmuir, 2010, 26, 215 CrossRef CAS PubMed.
- I. Francesco, C. Stefano and D. F. Rosa, J. Phys. Chem. C, 2008, 112, 13540 Search PubMed.
- A. J. Arduengo III, R. L. Harlow and M. J. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- S. Gründemann, A. Kovacevic, M. Albrecht, J. W. Faller and R. H. Crabtree, Chem. Commun., 2001, 2274 RSC.
- L. N. Appelhans, D. Zuccaccia, A. Kovacevic, A. R. Chianese, J. R. Miecznikowski, A. Macchioni, E. Clot, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2005, 127, 16299 CrossRef CAS PubMed.
- E. Kluser, A. Neels and M. Albrecht, Chem. Commun., 2006, 43, 4495 RSC.
- B. Cetinkaya, E. Cetinkaya, H. Küçükbay and R. Durmaz, Drug Res., 1996, 46, 821 CAS.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642 CrossRef CAS PubMed.
- J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978 CrossRef CAS PubMed.
- A. Fürstner, M. Alcarazo, V. Cesar and C. W. Lehmann, Chem. Commun., 2006, 28, 2176 RSC.
- H. D. Velázquez, Y. R. García, M. Vandichel, A. Madder and F. Verpoort, Org. Biomol. Chem., 2014, 12, 9350 Search PubMed.
- K. Oisaki, Q. Li, H. Furukawa, A. U. Czaja and O. Yaghi, J. Am. Chem. Soc., 2010, 132, 9262 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. G. Mavros, T. van Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2013, 135, 7418 CrossRef CAS PubMed.
- T. Weidner, J. E. Baio, A. Mundstock, C. Große, S. Karthäuser, C. Bruhn and U. Siemeling, Aust. J. Chem., 2011, 64, 1177 CrossRef CAS PubMed.
- M. C. Crudeen, et.al., Nat. Chem., 2014, 6, 409 CrossRef PubMed.
- K. Judai, S. Abbet, A. S. Wörz, A. M. Ferrari, L. Giordano, G. Pacchioni and U. Heiz, J. Mol. Catal. A: Chem., 2003, 199, 103 CrossRef CAS.
- R. W. Mayer, M. Melzer, M. Hävecker, A. Knop-Gericke, J. Urban, H. J. Freund and R. Schlögl, Catal. Lett., 2003, 86, 245 CrossRef CAS.
- M. A. Garcia, J. de la Venta, P. Crespo, J. L. Lopis, S. Penadés, E. Fernández and A. Hernando, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 241403 CrossRef.
- E. Guerrero, M. A. Muñoz-Márquez, M. A García, P. Crespo, E. Fernández-Pinel, A. Hernando and A. Fernández, Nanotechnology, 2008, 19, 175701 CrossRef CAS PubMed.
- H. H. Huang, F. Q. Yan, Y. M. Kek, C. H. Chew, G. Q. Xu, W. Ji, P. S. Oh and S. H. Tang, Langmuir, 1997, 13, 172 CrossRef CAS.
- N. A. Dhas, C. P. Raj and A. Gedanken, Chem. Mater., 1998, 10, 1446 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Nature, 1995, 376, 238 CrossRef CAS PubMed.
- B. Koun and C. M. Friend, Chem. Rev., 2007, 107, 2709 CrossRef PubMed.
- W. Jinlan, W. Guanghou and Z. Jijun, Chem. Phys. Lett., 2003, 380, 716–720 CrossRef PubMed.
- D. Y. Wu, B. Ren, Y. X. Jiang, X. Xu and Z. Q. Tian, J. Phys. Chem. A, 2002, 106, 9042 CrossRef CAS.
- D. Y. Wu, M. Hayashi, C. H. Chang, K. K. Liang and S. H. Lin, J. Chem. Phys., 2003, 118, 4073 CrossRef CAS PubMed.
- M. Prakash, K. Mathivon, D. M. Benoit, G. Chambaud and M. Hochlaf, Phys. Chem. Chem. Phys., 2014, 16, 12503 RSC.
- G. R. Souza, D. R. Christianson, F. I. Staquicini, M. G. Ozawa, E. Y. Snyder, R. L. Sidman, J. H. Miller, W. Arap and R. Pasqualini, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1215 CrossRef CAS PubMed.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS PubMed.
- G. R. Souza and J. H. Miller, J. Am. Chem. Soc., 2001, 123, 6734 CrossRef CAS.
- D. K. Christine, M. K. Kenneth and M. J. Natan, J. Phys. Chem., 1998, 102, 9404 CrossRef.
- L. R. Hirsch, J. B. Jackson, A. Lee, N. J. Halas and J. L West, Anal. Chem., 2003, 75, 2377 CrossRef CAS.
- S. R. Sershen, S. L. Westcott, N. J. Halas and J. L. West, J. Biomed. Mater. Res., 2000, 51, 293 CrossRef CAS.
- W. C. Shen, Biochim. Biophys. Acta, 1990, 1034, 122 CrossRef CAS.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Revision A.02, Inc., Wallingford, 2009 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS PubMed.
- Y. Akinaga and S. Ten-no, Chem. Phys. Lett., 2008, 462, 348 CrossRef CAS PubMed.
- S. Goel, A. K. A. Velizhanin, A. Piryatinski, S. Tretiak, S. A. Ivanov and J. Phy, Chem. Lett., 2010, 1, 927 CAS.
- E. J. Baerends, et al. Theoretical Chemistry, Organometallics, ADF 2010.01.SCM, Vrije Universiteit, Amsterdam, The Netherlands, 2012, 31, 4415, http://www.scm.com.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325 CrossRef CAS PubMed; K. Morokuma, Acc. Chem. Res., 1977, 10, 294 CrossRef.
- E. van Lenthe, A. E. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943 CrossRef CAS PubMed.
- C. Fonseca Guerra, J.-W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189 CrossRef PubMed.
- R. F. W. Bader, in Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed.
- C. F. Matta and R. J. Boyd, in The quantum theory of atoms in molecules, Wiley- VCH, Weinheim, Germany, 2007 Search PubMed.
- E. D. Glendening, A. E. Reed, J. A. Carpenter and F. Weinhold, NBO Version 31 Search PubMed.
- R. Tonner, G. Heydenrych and G. Frenking, Chem.–Asian. J., 2007, 2, 1555 CrossRef CAS PubMed.
- D. Y. Wu, M. Hayashi, Y. J. Shiu, K. K. Liang, C. H. Chang, Y. L. Yeh and S. H. Lin, J. Phys. Chem. A, 2003, 107, 9658 CrossRef CAS.
- T. Gießel, O. Schaff, R. Lindsay, P. Baumgärtel, M. Polcik, A. M. Bradshaw, A. Koebbel, T. McCabe, M. Bridge, D. R. Lloyd and D. P. Woodruff, J. Chem. Phys., 1999, 110, 9666 CrossRef PubMed.
- H. Häkkinen, Nat. Chem., 2012, 4, 443 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14578c |
| This journal is © The Royal Society of Chemistry 2015 |