
Corrosion inhibition of mild steel by an imidazolium ionic liquid compound: the effect of pH and surface pre-corrosion
Abstract
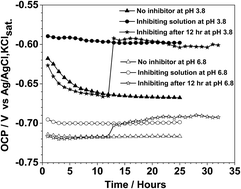
The corrosion inhibition performance of 1-decyl-3-methylimidazolium chloride (DMICL) on mild steel was investigated in a carbon dioxide-saturated NaCl solution at pH 3.8 and 6.8. Electrochemical and surface analysis techniques were used to characterize the corrosion process in blank and inhibiting solutions. Open circuit potential, polarization curves and impedance spectroscopy were recorded to analyze the inhibition behavior of DMICL for specimens with a well-polished surface and a pre-corroded surface. Scanning electron microscopy images showed that the specimen surfaces were well protected from uniform corrosion in the inhibition solution. The energy dispersive spectroscopy and X-ray photoelectron spectroscopy data indicated the presence of specific elements following a dissolution mechanism under different pH conditions. The electrochemical fitting data facilitated calculations of inhibition efficiency and accounted for the interfacial changes.
 Please wait while we load your content...
Please wait while we load your content...

