DOI:
10.1039/C5RA14480A
(Paper)
RSC Adv., 2015,
5, 101593-101598
Catalytic oxidation of cyclohexane by substituted metalloporphyrins: experimental and molecular simulation
Received
22nd July 2015
, Accepted 19th November 2015
First published on 20th November 2015
Abstract
A series of meso-substituted metal tetraphenylporphyrins were synthesized and characterized by UV-vis, FT-IR and elemental analysis. The experimental results show that cobalt tetra(4-nitrophonyl)porphyrin (Co–TNPP) gives better catalytic performance in cyclohexane oxidation. The KA oil (cyclohexanol and cyclohexanone) selectivity is 79.54% at the cyclohexane conversion of 10.43%, and the turnover number reaches to 0.93 × 104. Cyclohexane oxidation catalyzed by meso-substituted metalloporphyrins was studied by molecular simulation and experiment. The results of molecular simulation performed by Materials Studio show that the substituted electron-withdrawing groups in the same central metal porphyrins reduce the HOMO and LUMO energies. The metalloporphyrins with lower energy gap have stronger ability to activate the oxygen, the metalloporphyrin intermediates with lower energy gap have stronger ability to activate the cyclohexane.
1. Introduction
The oxidation of hydrocarbon is of great industrial and synthetic importance to obtain valuable products such as alcohols, ketones and carboxylic acids.1–3 Cyclohexane oxidation with air or dioxygen is one of these important processes to produce KA oil (cyclohexanol and cyclohexanone) or adipic acid, which are the raw materials for the production of nylon-6, nylon-6,6 and caprolactam.4–6 A large number of different types of catalysts have been tried in this reaction and get a certain progress.7–10 However, the cyclohexane conversion, KA oil selectivity and the harsh reaction conditions leave a lot to be desired. For the industrial cyclohexane catalytic oxidation process, the selectivity to KA oil is about 75% at the cyclohexane conversion of 8–10%.
Early in 1979, metalloporphyrins was used in catalytic oxidation by Groves, like cytochrome P-450, metalloporphyrins have been proven to be efficient oxidation catalysts and attracted many researchers' attention.11–14 It shows good activities and high selectivity in catalytic oxidation of hydrocarbons. The catalytic activity of metalloporphyrin is related to the number of substituents, the electron withdrawing ability of the substituents and the species of central metal ion.15–18 With the development of quantum chemistry, the frontier molecular orbital theory (FMO) proposed by Fukui became the most popular qualitative theory for description of chemical bonding rule.19,20 It holds that the capacity of molecular which gain or lose electrons is determined by the energy of HOMO and LUMO. The theory of FMO has been used in cyclohexane oxidation catalyzed by metalloporphyrins and it shows that the ΔEL–H and EHOMO have effects on the activity of the catalyst.21,22
In this paper, several substituted cobalt, manganese and nickel metalloporphyrins complexes (Co–D(p-Cl)D(p-OCH3)PP, Co–D(p-Br)D(p-Cl)PP, Co–TNPP, Mn–TNPP, Ni–TNPP) were synthesized and their catalytic performance in cyclohexane oxidation was studied. And the energy of molecular orbital of the catalysts and the formed intermediates were studied by using Dmol 3 program density functional computer software implemented in the Materials Studio.
2. Experimental
Reagents
All reagents and solvents used were analytical grade and were obtained from Aladdin Reagent. No impurities were found in cyclohexane by GC analysis before use.
Synthesis of meso-substituted metalloporphyrins
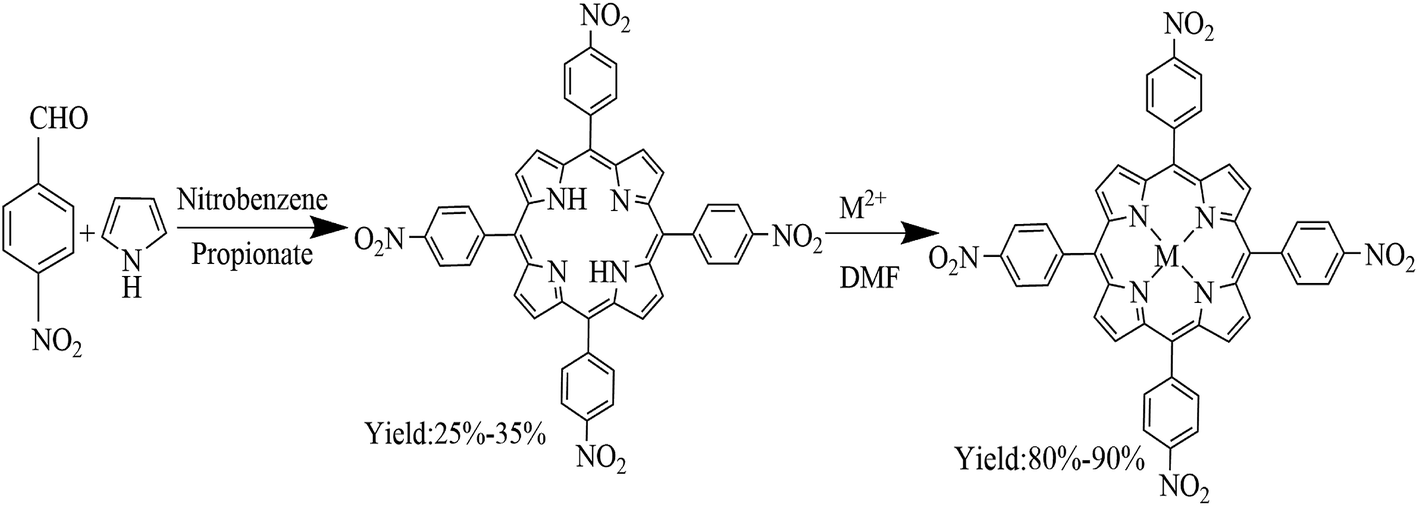
Co–D(p-Cl)D(p-OCH3)PP and Co–D(p-Br)D(p-Cl)PP) were synthesized by referring to the following procedures.17,23–25 Co–TNPP, Ni–TNPP and Mn–TNPP was synthesized according to the following procedures26,27 (Scheme 1). 1.24 mmol tetra(4-nitrophonyl)porphyrin (TNPP) and 6.21 mmol cobalt acetate (or nickel acetate or manganese acetate) was added to the 100 mL DMF solution and heated at reflux, thin layer chromatography was used to monitor the evolution of the reaction. After the reaction finished, the mixture was filtered. The metalloporphyrins was purified by column chromatography (neutral aluminum oxide, CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol = 10:1.5).
:methanol = 10:1.5).
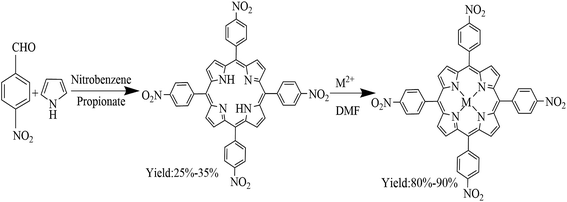 |
| | Scheme 1 Synthesis of porphyrin and metalloporphyrin. | |
Catalysts characterization
The porphyrins and metalloporphyrins were characterized by 1HNMR (Bruker Avance, 400 MHz, CDCl3).17 the TNPP, Mn–TNPP, Ni–TNPP and Co–TNPP were characterized by UV-vis (UV-2550 spectrophotometry), FT-IR (Nicolet iS10 spectrometer) and elemental analysis (Vario EL III).
Procedures for the catalytic test
Cyclohexane (15.82 g) and catalyst (1.8 mg) were added in a 50 mL autoclave reactor with a magnetic stirrer in the absence of solvent. The reactor was sealed and the gas in autoclave was replaced with oxygen for two times. Then the autoclave was heated to the setting temperature. After the reaction, the reactor was cooled to the ambient temperature and the mixture was dissolved in ethanol and the catalyst was removed by filtration. The products of the reaction mixture were identified by GC-MS and LC-MS. The acids in the product are mainly the succinic acid, glutaric acid and adipic acid. The products of cyclohexanol and cyclohexanone were analyzed by GC using chlorobenzene as the internal standard, and the cyclohexyl hydroperoxide and by-products (acid and ester) were analyzed by triphenylphosphine and chemical titration.
3. Results and discussion
Characterization of the porphyrins and metalloporphyrins
The results of elemental analysis of the TNPP, Mn–TNPP and Co–TNPP are shown in Table 1. It can be seen from Table 1 that the elemental analysis results of TNPP, Mn–TNPP, Co–TNPP are closely coincident with the theoretical value, which means that the porphyrins and metalloporphyrins were successfully synthesized, and the purity meet the requirements.
Table 1 Results of elemental analysis
| Porphyrin |
C% |
H% |
N% |
| Theory |
Exp. |
Theory |
Exp. |
Theory |
Exp. |
| TNPP |
66.50 |
66.32 |
3.27 |
3.18 |
14.10 |
14.01 |
| Mn–TNPP |
62.32 |
62.03 |
2.83 |
2.76 |
13.22 |
13.11 |
| Co–TNPP |
62.05 |
61.92 |
2.82 |
2.82 |
13.16 |
12.93 |
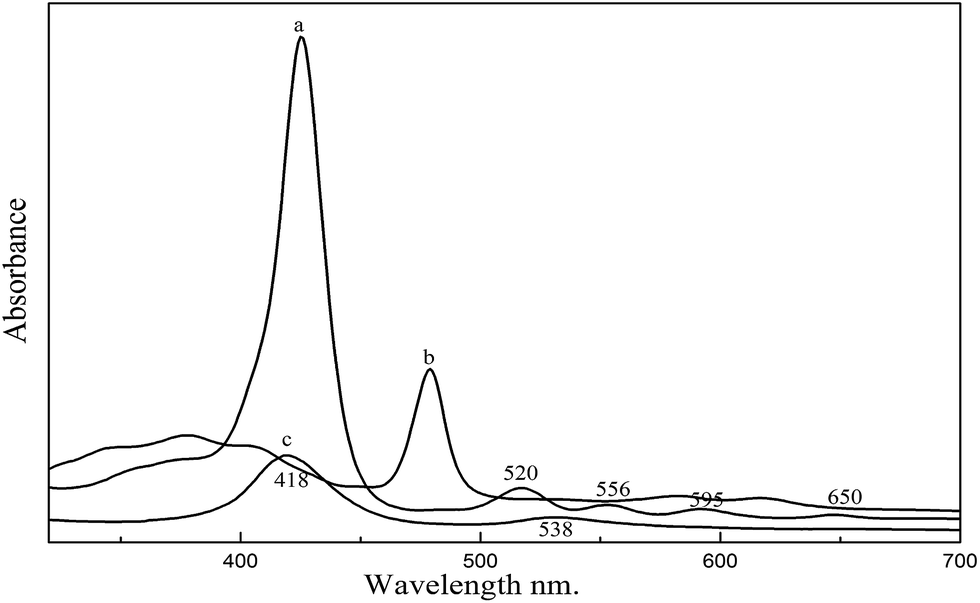
UV-vis spectra of the TNPP, Mn–TNPP and Co–TNPP are shown in Fig. 1. There are one Soret band and four Q bands in spectra (a), which are characteristic absorption peaks of porphyrins. Compared with the TNPP, the spectra of Mn–TNPP is blue-shifted, the reason is that the Mn(III) ion inserts into the free base porphyrin and forms the manganese metalloporphyrin,28 and the numbers of Q bands decrease from four to two peaks resulted from alteration in the micro-symmetry of the porphyrin macrocycle from D2h to D4h.29 For the UV spectra of Co–TNPP, the Soret band of Co–TNPP is red-shifted to 418 nm, and the Q band at 520 nm, 556 nm, 595 nm and 650 nm disappear and the new Q band forms at 538 nm. Such phenomenon may occur when the porphyrin is changed to metal porphyrin.30
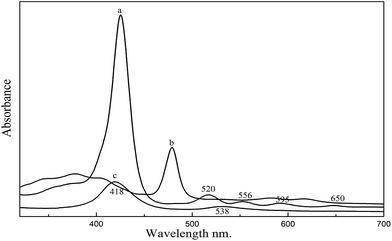 |
| | Fig. 1 UV-vis spectra of porphyrins and metalloporphyrins. (a) TNPP, (b) Mn–TNPP, (c) Co–TNPP. | |
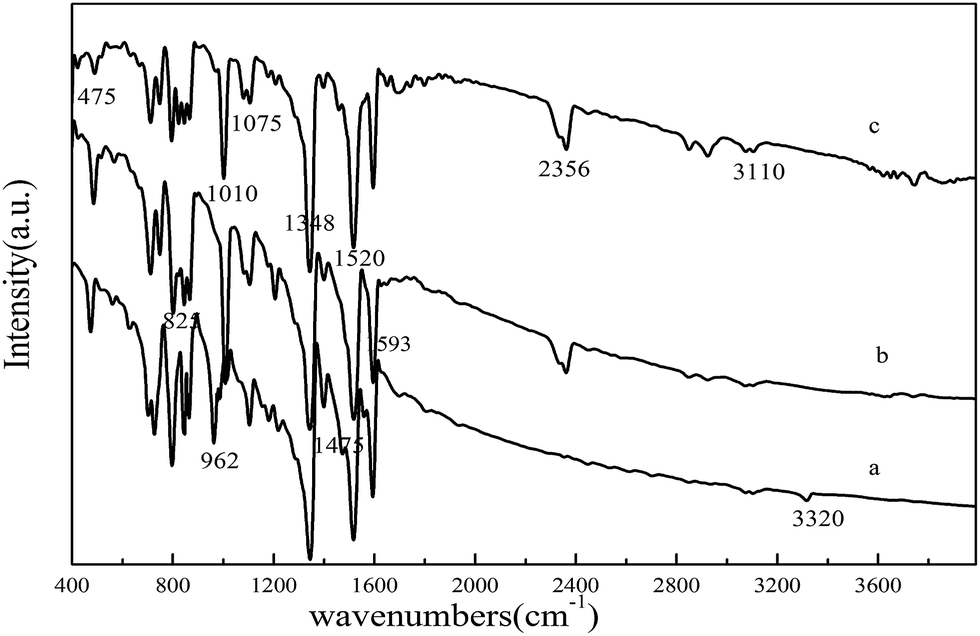
The FT-IR spectra of TNPP, Mn–TNPP and Co–TNPP are shown in Fig. 2. There are two peaks of 962 cm−1 and 3320 cm−1 in TNPP, which is the bending and stretching vibrations of N–H in the pyrrole ring.31 However, these two peaks disappear in the corresponding metal porphyrins, it means that the bonds of N–H in the pyrrole have been replaced by the Co–N and Mn–N, this indicates that the metal ions successfully embedded into the porphyrin ring. The peaks of 1348 cm−1 and 1520 cm−1 in the spectra are assigned to the symmetric and asymmetric stretching vibration of the –NO2 group, and the peak at 825 cm−1 shows that the nitro group is in the para position of the benzene ring.32,33 The vibration peaks of phenyl in the porphyrin appear at 1593 cm−1 and 3110 cm−1. When metal ions anchored into the porphyrin ring and formed a new bond, the new M–N bond would affect the vibration frequency of Cα–Cβ, Cα–Cm and Cβ–H which is close to the M–H bond, and then some metal sensitive zones form.34 Compared with the FT-IR spectra of porphyrin, the metal sensitive zone of the metalloporphyrin appeared at 475 cm−1, 1010 cm−1, 1075 cm−1, 1475 cm−1 and 2356 cm−1, it indicates that we the corresponding metalloporphyrins were successfully synthesized.
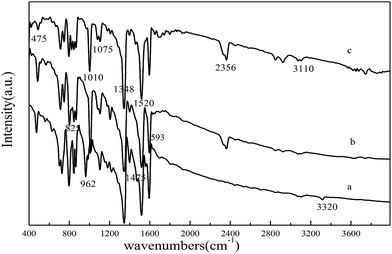 |
| | Fig. 2 FT-IR spectra of (a) TNPP, (b) Mn–TNPP and (c) Co–TNPP. | |
Catalytic performance
Several different meso-substituted metal tetraphenylporphyrins were synthesized and applied in catalytic oxidation of cyclohexane.10,14,17 In this paper, meso-substituted metal nitrophonylporphyrins were prepared and the catalytic performance of these meso-substituted metalloporphyrins are showed in Table 2. It can be seen from Table 2 that the species of substituents have great effects on the catalytic performance. Co–TNPP presents the highest cyclohexane conversion and KA oil yield under the same reaction conditions (Table 2 entries 1–3). It has shown that the reaction rate of cyclohexane hydroxylation is speeded up by electron-withdrawing groups in the substituted mentalloporphyrins.35,36 The electron-withdrawing ability are as follows: –NO2 > –Cl > –Br > –OCH3, hence, the cobalt tetra(4-nitrophonyl)porphyrin (Co–TNPP) shows better catalytic performance. However, there is a special case when the substitutes were substituted in the propionate side chains of metalloporphyrin in the oxidation of toluene.37 The different central metal in the metalloporphyrin has different ionization potential.38–40 It can be seen from Table 2 (entries 4–6) that the cobalt metalloporphyrin shows the best catalytic activity among these metalloporphyrins with same substituents, it gives the highest cyclohexane conversion, KA oil yield and the turnover number. And it is in agreement with the reported literature.41
Table 2 Results of cyclohexane oxidation catalyzed by different metalloporphyrins
| Entry |
Catalyst |
Conversion (%) |
K–A selectivity (%) |
K/A (mol ratio) |
TONc (×104) |
| Reaction conditions: cyclohexane 0.188 mol, catalysts 1.8 mg, 155 °C for 1 h, oxygen pressure 1.0 MPa. Reaction conditions: cyclohexane 0.188 mol, catalysts 1.8 mg, 150 °C for 2 h, oxygen pressure 1.0 MPa. The turnover number (TON) is the value calculated by mole product (ketone + alcohol + acid + ester)/mol metalloporphyrin. |
| 1a |
Co–D(p-Cl)D(p-OCH3)PP |
7.20 |
78.05 |
0.64 |
0.59 |
| 2b |
Co–D(p-Br)D(p-Cl)PP |
9.23 |
74.54 |
0.61 |
0.85 |
| 3a |
Co–TNPP |
9.54 |
77.94 |
0.58 |
0.85 |
| 4b |
Co–TNPP |
11.54 |
74.25 |
0.83 |
1.03 |
| 5b |
Mn–TNPP |
9.76 |
74.70 |
0.56 |
0.86 |
| 6b |
Ni–TNPP |
9.23 |
72.67 |
0.63 |
0.81 |
Molecular simulation calculation
During the process of cyclohexane oxidation with dioxygen catalyzed by metalloporphyrins, the mechanism of cyclohexane oxidation was studied and it suppose to form the intermediates of peroxo-metal anion (metalloporphyrins![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).42,43 The molecular simulation calculation was carried out in order to obtain the information of the ability of different metalloporphyrins to form the above mentioned intermediates. The structure of metalloporphyrins were optimized by the Dmol3 module of Material Studio 6.1, and then the optimized structures were used to calculate HOMO and LUMO energies of metalloporphyrin complexes and the density mixing (charge 0.2, spin 0.5) by the BLYP general-gradient approximation (GGA) in conjunction with the double-numerical basis set. The calculation results are shown in Table 3. As to the metalloporphyrins with the same central metal ion, the HOMO and LUMO energies decrease with the increment of the electron-withdrawing substituents (entries 1–3 in Table 3), and the electron-withdrawing substituent could reduce negative charges, for the stronger of the electron-withdrawing substituents, it is easier to reduce the charge density on the porphyrin ring, and improve the ability of the central metal ion binding of oxygen. As to the metalloporphyrins with the same substituents, the species of central metal ion have great influence on the energy of HOMO and have a little effect on the energy of LUMO. It can be found that the energy gap of manganese metalloporphyrins is approximately 0.5 eV, which is much lower than that of cobalt and nickel metalloporphyrins (entries 3–5). It also can be seen that the energy of HOMO(Mn–TNPP) is almost the same as the energy of LUMO(O2), and the energy of LUMO(Mn–TNPP) is most closely to the energy of HOMO(O2), this means that it is easier for the electrons to flow between the Mn–TNPP and oxygen atoms than that of Co–TNPP and Ni–TNPP.44,45 Hence, the Mn–TNPP have better property of activating oxygen. Meanwhile, compared with the LUMO orbit of Co–TNPP and Ni–TNPP, the LUMO orbit of Mn–TNPP contains the highest proportion of the a2u orbit, which is beneficial to coordinate with oxygen molecule in the mode of side-on.46 The formation of porphyrin intermediates may be that the central metal MIII of the (por)MIII captures an electronic and forms the (por)MII, and the divalent metal reacts with oxygen molecule to form the (por)MIII–OO−, then the intermediate of (por)MIII–OO− interacts with the (por)MII and generates an peroxide of (por)MIII–O–O–(por)MIII, the intermediates of (por)MIVO may be formed after breakage of the peroxide bond.47
O).42,43 The molecular simulation calculation was carried out in order to obtain the information of the ability of different metalloporphyrins to form the above mentioned intermediates. The structure of metalloporphyrins were optimized by the Dmol3 module of Material Studio 6.1, and then the optimized structures were used to calculate HOMO and LUMO energies of metalloporphyrin complexes and the density mixing (charge 0.2, spin 0.5) by the BLYP general-gradient approximation (GGA) in conjunction with the double-numerical basis set. The calculation results are shown in Table 3. As to the metalloporphyrins with the same central metal ion, the HOMO and LUMO energies decrease with the increment of the electron-withdrawing substituents (entries 1–3 in Table 3), and the electron-withdrawing substituent could reduce negative charges, for the stronger of the electron-withdrawing substituents, it is easier to reduce the charge density on the porphyrin ring, and improve the ability of the central metal ion binding of oxygen. As to the metalloporphyrins with the same substituents, the species of central metal ion have great influence on the energy of HOMO and have a little effect on the energy of LUMO. It can be found that the energy gap of manganese metalloporphyrins is approximately 0.5 eV, which is much lower than that of cobalt and nickel metalloporphyrins (entries 3–5). It also can be seen that the energy of HOMO(Mn–TNPP) is almost the same as the energy of LUMO(O2), and the energy of LUMO(Mn–TNPP) is most closely to the energy of HOMO(O2), this means that it is easier for the electrons to flow between the Mn–TNPP and oxygen atoms than that of Co–TNPP and Ni–TNPP.44,45 Hence, the Mn–TNPP have better property of activating oxygen. Meanwhile, compared with the LUMO orbit of Co–TNPP and Ni–TNPP, the LUMO orbit of Mn–TNPP contains the highest proportion of the a2u orbit, which is beneficial to coordinate with oxygen molecule in the mode of side-on.46 The formation of porphyrin intermediates may be that the central metal MIII of the (por)MIII captures an electronic and forms the (por)MII, and the divalent metal reacts with oxygen molecule to form the (por)MIII–OO−, then the intermediate of (por)MIII–OO− interacts with the (por)MII and generates an peroxide of (por)MIII–O–O–(por)MIII, the intermediates of (por)MIVO may be formed after breakage of the peroxide bond.47
Table 3 HOMO and LUMO energies and HOMO–LUMO gap for metalloporphyrins
| Entry |
Complex |
HOMO (Ha) |
LUMO (Ha) |
Energy gap (eV) |
ΔE(L(P)–H(O2)) (eV) |
ΔE(L(O2)–H(P)) (eV) |
| 1 |
Co–D(p-Cl)D(p-OCH3)PP |
−0.17433 |
−0.11176 |
1.70253 |
3.81348 |
0.15863 |
| 2 |
Co–D(p-Br)PD(p-Cl)P |
−0.18432 |
−0.12037 |
1.74008 |
3.57920 |
0.43046 |
| 3 |
Co–TNPP |
−0.20615 |
−0.14124 |
1.76634 |
3.01140 |
1.02464 |
| 4 |
Mn–TNPP |
−0.16806 |
−0.14893 |
0.52061 |
2.80231 |
−0.01200 |
| 5 |
Ni–TNPP |
−0.19862 |
−0.13796 |
1.65078 |
3.10073 |
0.81974 |
| 6 |
O2 |
−0.25191 |
−0.16850 |
2.26959 |
|
|
The HOMO and LUMO orbits energies of metalloporphyrinsO and cyclohexane were calculated in order to obtain the information of the cyclohexane activation ability of the intermediates of metalloporphyrinsO. As to the cobalt porphyrins intermediates with different substituents, the energies of HOMO orbit decrease with the increment of the electron-withdrawing substituents (entries 1–3 in Table 4). And the substituent of –Br improves the LUMO energy of Co–D(p-Br)PD(p-Cl)PO. As to the metalloporphyrinsO with different central metal ions, the energy gap of Co–TNPPO is much lower than Mn–TNPPO and Ni–TNPPO, this means that it is more easier for the Co–TNPPO to activate the inert cyclohexane, it can also be seen that the HOMO energy of cyclohexane is most closely to the LUMO energy of Co–TNPPO, and the LUMO energy of cyclohexane is most closely to the HOMO energy of Co–TNPP, this means that it is more easier for the electrons of cyclohexane to flow to Co–TNPPO and produce cyclohexyl radical. The results of experiment indicate that Co–TNPP presents the best catalytic performance among these metalloporphyrins catalysts under the same reaction condition with dioxygen, and the catalytic activity is mainly concerned with the ability of the metalloporphyrinsO intermediates to activate the inert cyclohexane. The activation of cyclohexane may be that the intermediate of (por)MIVO interacts with cyclohexane and capture a hydrogen atom from cyclohexane to form the cyclohexyl radical and (por)MIII–OH. On one hand, the cyclohexyl radical could react with oxygen molecule to form cyclohexyl peroxy radicals, cyclohexanol and cyclohexanone, on the other hand, the cyclohexyl radical could react with (por)MIII–OH and the –OH transfer from the central metal ion to cyclohexyl radical to form cyclohexanol, and the central metal is reduced to (por)MII.48
Table 4 HOMO and LUMO energies and HOMO–LUMO gap for metalloporphyrinsO
| Entry |
Complex |
HOMO (Ha) |
LUMO (Ha) |
Energy gap (eV) |
ΔE(L(P)–H(C)) (eV) |
ΔE(L(C)–H(P)) (eV) |
| 1 |
Co–D(p-Cl)PD(p-OCH3)PO |
−0.14467 |
−0.14363 |
0.02841 |
2.90393 |
5.57884 |
| 2 |
Co–D(p-Br)PD(p-Cl)PO |
−0.16557 |
−0.11543 |
1.36409 |
3.67106 |
6.14739 |
| 3 |
Co–TNPPO |
−0.17102 |
−0.17013 |
0.02400 |
2.18270 |
6.29631 |
| 4 |
Mn-TNPPO |
−0.20724 |
−0.16080 |
1.26337 |
2.43659 |
7.28163 |
| 5 |
Ni–TNPPO |
−0.19086 |
−0.14330 |
1.29417 |
2.91298 |
6.83604 |
| 6 |
Cyclohexane |
−0.25035 |
0.06036 |
8.45436 |
|
|
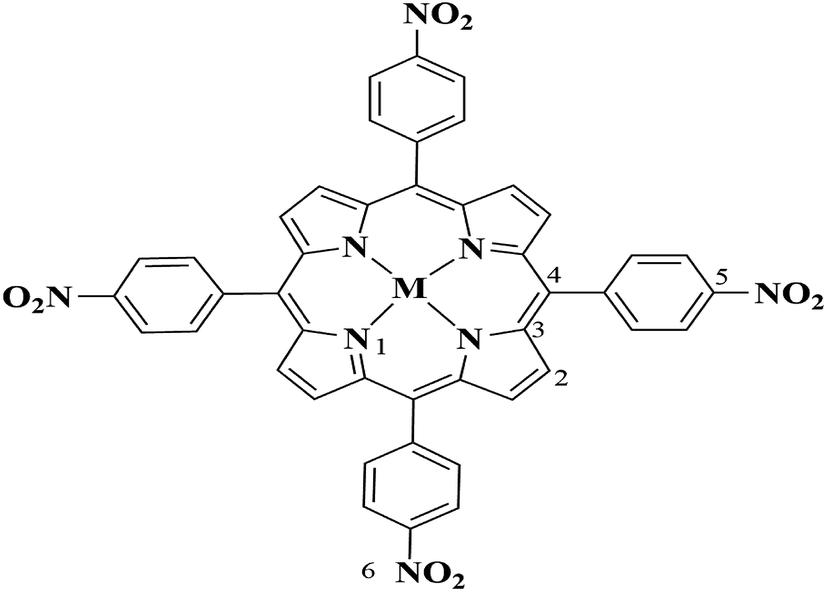
As to the same central metal ions, the metalloporphyrins with stronger electron-withdrawing substituents are easier to adsorb more charges from the porphyrin ring, and thus increase the positive charge of the central metal ion and the activity of metalloporphyrins.46 For the metalloporphyrins with the same substituents, the atomic of nitro-substituted metal porphyrins are marked in the Fig. 3, and the Mulliken atomic charges of the M(II) and M(IV) oxo states are shown in Table 5, the manganese ions have the highest positive charge (entry 1–3), which indicates that the Mn–TNPP is more easy to activate the oxygen molecule. The charge of the central metal ions changes among the metalloporphyrin intermediates, the positive charge of cobalt ion increases greatly, while the positive charge of manganese ion and the nickel ion increase a little, hence, the positive ion of cobalt in metalloporphyrin intermediates are the highest, and the M(IV) oxo states of cobalt metalloporphyrin is more easy to activate cyclohexane. The charges of the other elements does not change greatly.
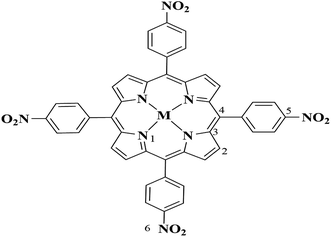 |
| | Fig. 3 The marked nitro-substituted metalloporphyrin. | |
Table 5 Mulliken atomic charges of metalloporphyrins and its intermediates
| Entry |
Complex |
M |
N1 |
N6 |
C2 |
C3 |
C4 |
C5 |
| 1 |
Co–TNPP |
0.601 |
−0.503 |
0.346 |
−0.252 |
0.256 |
−0.015 |
0.134 |
| 2 |
Mn–TNPP |
0.754 |
−0.555 |
0.344 |
−0.259 |
0.264 |
−0.039 |
0.111 |
| 3 |
Ni–TNPP |
0.595 |
−0.514 |
0.346 |
−0.247 |
0.259 |
−0.011 |
0.145 |
| 4 |
Co–TNPPO |
0.761 |
−0.477 |
0.347 |
−0.250 |
0.263 |
−0.006 |
0.135 |
| 5 |
Mn–TNPPO |
0.757 |
−0.548 |
0.344 |
−0.256 |
0.266 |
−0.036 |
0.111 |
| 6 |
Ni–TNPPO |
0.605 |
−0.507 |
0.345 |
−0.250 |
0.256 |
−0.009 |
0.145 |
Cyclohexane oxidation over Co–TNPP
The results of cyclohexane oxidation over Co–TNPP under different reaction conditions are shown in Table 6. The effects of reaction temperature are shown in entries 1–4 in Table 6. At lower temperature of 140 °C, the cyclohexane conversion is 6.48 and the selectivity to KA oil is 85.63%, the selectivity to KA oil is obviously decreased with the increment of temperature, the reason may be that the product of cyclohexanone and cyclohexanol are more-readily oxidizable than cyclohexane. Entries 2, 5 and 6 in Table 5 show the effects of reaction time on cyclohexane oxidation catalyzed by Co–TNPP. The cyclohexane conversion changes little from 1 h to 3 h, while the selectivity to KA oil decreases from 79.54% to 73.87%, it possibly because that cyclohexane oxidation is a radical reaction, after the chain initiation reaction, the main reaction would be completed in a short time, and it has no significant impact to the conversion of this reaction with the time extending under a relatively low temperature and pressure conditions, and the reduction of the selectivity may be attributed to further oxidation of cyclohexanone and cyclohexanol. Entries 2, 7 and 8 in Table 6 show the effects of reaction pressure on cyclohexane oxidation catalyzed by Co–TNPP. The cyclohexane conversion increases markedly and the selectivity to KA oil decreases significantly with the increment of dioxygen pressure. Meanwhile, the TON and K/A mol ratio increase with reaction pressure.
Table 6 Results of cyclohexane oxidation over Co–TNPP under different reaction conditions
| Entry |
Cov. (%) |
KA sel. (%) |
K/A (mol ratio) |
TONd (×104) |
| Reaction conditions: cyclohexane (0.188 mol), catalyst (Co–TNPP: 1.8 mg), time (1.0 h). Oxygen pressure (1.0 MPa), temperature (entry 1–4: 140 °C, 150 °C, 155 °C, 160 °C). Reaction conditions: cyclohexane (0.188 mol), catalyst (Co–TNPP: 1.8 mg), oxygen pressure 1.0 MPa, temperature (150 °C), time (entry 5–6, 2.0 h, 3.0 h). Reaction conditions: cyclohexane (0.188 mol), catalyst (Co–TNPP: 1.8 mg), temperature (150 °C), time (1.0 h). Oxygen pressure (entry 7–8, 1.5 MPa, 2.0 MPa). The turnover number (TON) = mol product (ketone + alcohol + acid + ester)/mol catalyst. |
| 1a |
6.48 |
85.63 |
0.66 |
0.57 |
| 2a |
10.43 |
79.54 |
0.65 |
0.93 |
| 3a |
9.54 |
77.94 |
0.58 |
0.85 |
| 4a |
8.31 |
77.58 |
0.57 |
0.74 |
| 5b |
11.54 |
74.25 |
0.83 |
1.03 |
| 6b |
11.45 |
73.87 |
0.82 |
1.02 |
| 7c |
17.66 |
61.02 |
1.12 |
1.58 |
| 8c |
19.71 |
59.74 |
1.28 |
1.76 |
4. Conclusion
A series of meso-substituted metalloporphyrins were prepared, characterized and applied in cyclohexane oxidation with dioxygen. Molecular simulation calculation was carried out by using Material Studios 6.1. The calculation results show that the electron-withdrawing groups may reduce the energies of HOMO and LUMO orbits. The metalloporphyrins with lower energy gap have stronger ability to activate the oxygen. The metalloporphyrinO intermediates with lower energy gap have stronger ability to activate the cyclohexane. The catalytic activity of the metalloporphyrins in cyclohexane oxidation with dioxygen is mainly concerned with the ability of the metalloporphyrinsO intermediates to activate the inert cyclohexane.
Acknowledgements
This work was supported by the NSFC (21276218, 21306160) SRFDP (20124301110007), and the project of Xiangtan University (2015SEP05).
References
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- M. Sun, J. Zhang, P. Putaj, V. Caps, F. Lefebvre, J. Pelletier and J. M. Basset, Chem. Rev., 2013, 114, 981–1019 CrossRef PubMed.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed.
- R. Raja, G. Sankar and J. M. Thomas, J. Am. Chem. Soc., 1999, 121, 11926–11927 CrossRef CAS.
- R. Jevtic, P. A. Ramachandran and M. P. Dudukovic, Ind. Eng. Chem. Res., 2009, 48, 7986–7993 CrossRef CAS.
- Z. Gui, W. Cao, L. Chen and Z. Qi, Catal. Commun., 2015, 64, 58–61 CrossRef CAS.
- P. Srinivas and M. Mukhopadhyay, Ind. Eng. Chem. Res., 1997, 36, 2066–2074 CrossRef CAS.
- H. Guan, Y. A. Guo, Z. Hong, S. K. Zhao, S. Y. Liu, A. P. Wang and J. F. Wei, J. Mol. Catal. A: Chem., 2007, 273, 144–148 CrossRef.
- Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2010, 1, 2–6 CrossRef.
- Y. Xie, F. Zhang, P. Liu, F. Hao and H. A. Luo, J. Mol. Catal. A: Chem., 2014, 386, 95–100 CrossRef CAS.
- J. T. Groves and R. M. Dias, J. Am. Chem. Soc., 1979, 101, 1032–1033 CrossRef CAS.
- D. Mansuy, C. R. Chim., 2007, 10, 392–413 CrossRef CAS.
- M. Zhao, S. Ou and C. D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed.
- Y. Xie, F. Zhang, P. Liu, F. Hao and H. Luo, Ind. Eng. Chem. Res., 2015, 54, 2425–2430 CrossRef CAS.
- I. Hermans, P. Jacobs and J. Peeters, Chemistry, 2007, 13, 754–761 CrossRef CAS PubMed.
- W. P. To, Y. Liu, T. C. Lau and C. M. Che, Chem.–Eur. J., 2013, 19, 5654–5664 CrossRef CAS PubMed.
- Y. Xie, F. Zhang, P. Liu, F. Hao and H. A. Luo, Can. J. Chem., 2014, 92, 49–53 CrossRef CAS.
- Y. Zhang, W. Dai, G. Wu, N. Guan and L. Li, Chin. J. Catal., 2014, 35, 279–285 CrossRef CAS.
- K. Fukui, T. Yonezawa and H. Shingu, J. Chem. Phys., 1952, 20, 722–725 CrossRef CAS.
- J. Anderson, J. Melin and P. Ayers, Conceptual density-functional theory for general chemical reactions, including those that are neither charge- nor frontier-orbital-controlled. 1. Theory and derivation of a general-purpose reactivity indicator - ResearchGate, J. Chem. Theory Comput., 2007, 3, 358–374 CrossRef CAS.
- Y.-H. Zhang, Y.-B. She, R.-G. Zhong, X.-T. Zhou and H.-B. Ji, Acta Chim. Sin., 2004, 62, 2228–2232 CAS.
- L. J. Bartolotti and P. W. Ayers, J. Phys. Chem. A, 2005, 109, 1146–1151 CrossRef CAS PubMed.
- R. Naik, P. Joshi, N. V. S. P. Kaiwar and R. K. Deshpande, Tetrahedron, 2003, 59, 2207–2213 CrossRef CAS.
- J. S. Lindsey, Acc. Chem. Res., 2010, 43, 300–311 CrossRef CAS PubMed.
- A. R. Silva, T. Mourão and J. o Rocha, Catal. Today, 2013, 203, 81–86 CrossRef CAS.
- B. Zhu, Z. Xu and Y. Xu, Chin. J. Appl. Chem., 1999, 16, 68–70 CAS.
- H. Guan, Z. C. Luo, X. Feng, C. Xuan, Y. A. Guo and Y. X. Jiang, J. Mol. Catal. A: Chem., 2011, 340, 60–64 CrossRef.
- P. Bhyrappa, J. K. Young, J. S. Moore and K. S. Suslick, J. Am. Chem. Soc., 1996, 118, 5708–5711 CrossRef CAS.
- W. Zheng, N. Shan, L. Yu and X. Wang, Dyes Pigm., 2008, 77, 153–157 CrossRef CAS.
- G. K. B. Ferreira, G. S. Machado, R. R. Ribeiro, K. J. Ciuffi, G. P. Ricci, J. A. Marques and S. Nakagaki, J. Mol. Catal. A: Chem., 2013, 378, 263–272 CrossRef CAS.
- A. T. Dubis, S. a. J. Grabowski, D. B. Romanowska, T. Misiaszek and J. Leszczynski, J. Phys. Chem. A, 2002, 106, 10613–10621 CrossRef CAS.
- T. S. Kurtikyan, A. A. Hovhannisyan, M. E. Hakobyan, J. C. Patterson, A. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2007, 129, 3576–3585 CrossRef CAS PubMed.
- T. S. Kurtikyan, V. A. Hayrapetyan, M. M. Mehrabyan and P. C. Ford, Inorg. Chem., 2014, 53, 11948–11959 CrossRef CAS PubMed.
- J. L. Garate-Morales, F. S. Tham and R. A. Christopher, Inorg. Chem., 2007, 46, 1514–1516 CrossRef CAS PubMed.
- A. Maldotti, C. Bartocci, G. Varani, A. Molinari, P. Battioni and D. Mansuy, Inorg. Chem., 1996, 35, 1126–1131 CrossRef CAS PubMed.
- M. K. Wong, N. W. Chung, L. He, X. C. Wang, Z. Yan, Y. C. Tang and D. Yang, J. Cheminf., 2003, 34, 6321–6328 Search PubMed.
- B. Hu, C. Sun, Q. Deng and Z. Liu, J. Inclusion Phenom. Macrocyclic Chem., 2013, 76, 345–352 CrossRef CAS PubMed.
- H. He, Y. Lei, C. Xiao, D. Chu, R. Chen and G. Wang, J. Phys. Chem. C, 2012, 116, 16038–16046 CAS.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- J. Tong, L. Bo, X. Cai, H. Wang, Q. Zhang and L. Su, Ind. Eng. Chem. Res., 2014, 53, 10294–10300 CrossRef CAS.
- C. C. Guo, M. F. Chu, L. Qiang, L. Yang, D. C. Guo and X. Q. Liu, Appl. Catal., A, 2003, 246, 303–309 CrossRef CAS.
- O. Zakharieva, A. X. Trautwein and C. Veeger, Biophys. Chem., 2000, 88, 11–34 CrossRef CAS.
- H. Noack, V. Georgiev, M. R. A. Blomberg, P. E. M. Siegbahn and A. J. Johansson, Inorg. Chem., 2011, 50, 1194–1202 CrossRef CAS PubMed.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1970, 8, 17–22 Search PubMed.
- T. Vangberg, R. Lie and A. Ghosh, J. Am. Chem. Soc., 2002, 124, 8122–8130 CrossRef CAS PubMed.
- Y. Y. Cao Meijuan, F. U. Haiyan and S. H. E. Yuanbin, CIESC J., 2013, 64, 88–97 Search PubMed.
- H. Guan, S. Y. Liu, Y. A. Guo, A. P. Wang, L. Jin and C. C. Cai, Appl. Catal., A, 2009, 358, 173–179 CrossRef.
- L. Weber, R. Hommel, J. Behling, G. Haufe and H. Hennig, J. Am. Chem. Soc., 1994, 116, 2400–2408 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.